

Invasion emerges from cancer cell adaptation to competitive microenvironments: quantitative predictions from multiscale mathematical models
- PMID: 18524624
- PMCID: PMC3789515
- DOI: 10.1016/j.semcancer.2008.03.018
Invasion emerges from cancer cell adaptation to competitive microenvironments: quantitative predictions from multiscale mathematical models
Abstract
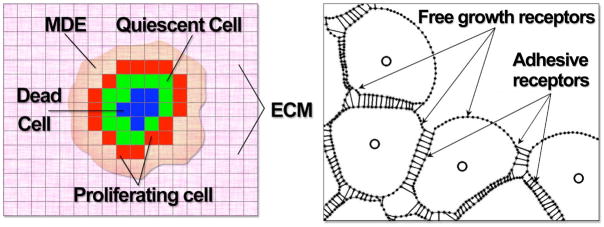
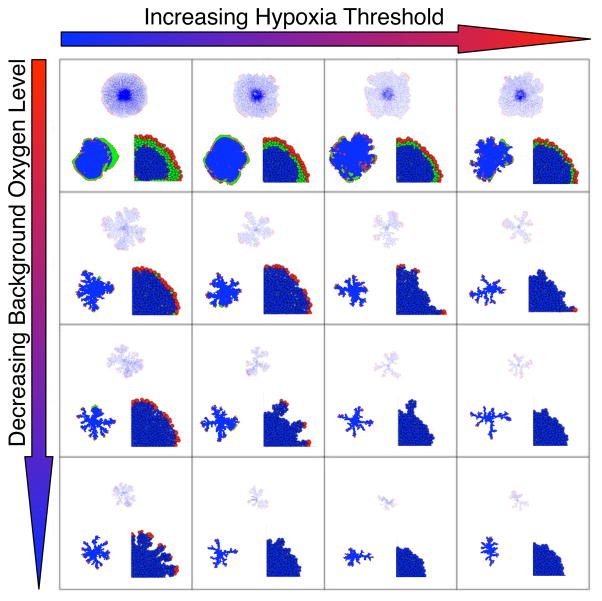
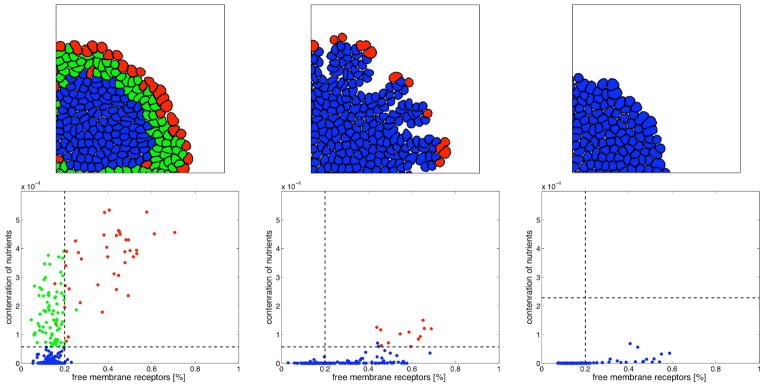
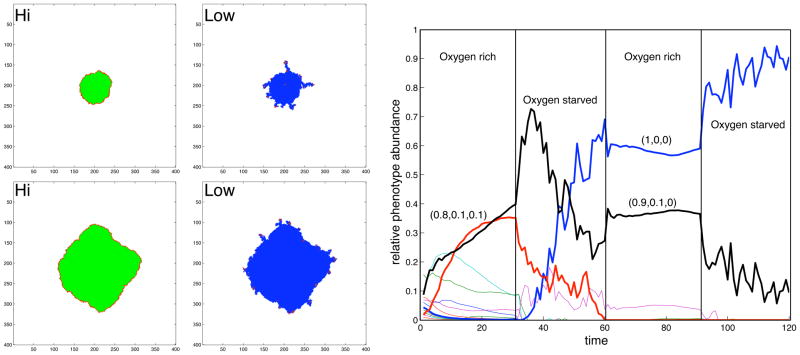
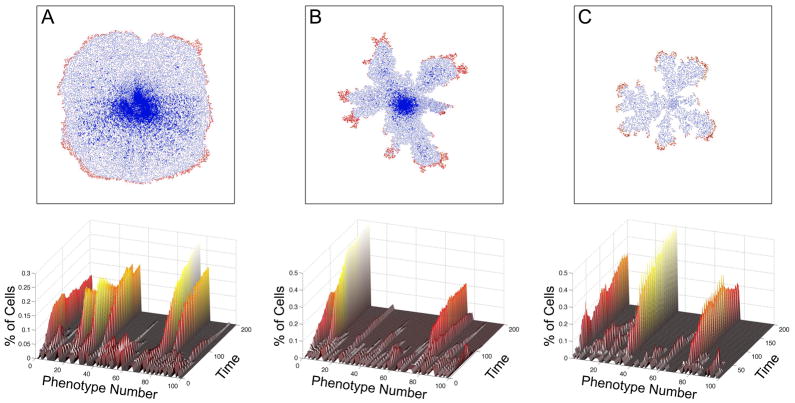
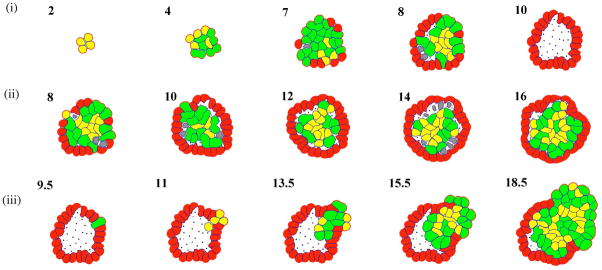
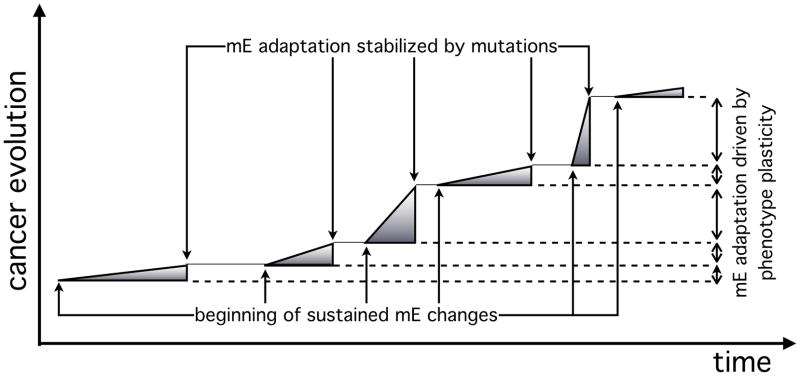
In this review we summarize our recent efforts using mathematical modeling and computation to simulate cancer invasion, with a special emphasis on the tumor microenvironment. We consider cancer progression as a complex multiscale process and approach it with three single-cell-based mathematical models that examine the interactions between tumor microenvironment and cancer cells at several scales. The models exploit distinct mathematical and computational techniques, yet they share core elements and can be compared and/or related to each other. The overall aim of using mathematical models is to uncover the fundamental mechanisms that lend cancer progression its direction towards invasion and metastasis. The models effectively simulate various modes of cancer cell adaptation to the microenvironment in a growing tumor. All three point to a general mechanism underlying cancer invasion: competition for adaptation between distinct cancer cell phenotypes, driven by a tumor microenvironment with scarce resources. These theoretical predictions pose an intriguing experimental challenge: test the hypothesis that invasion is an emergent property of cancer cell populations adapting to selective microenvironment pressure, rather than culmination of cancer progression producing cells with the "invasive phenotype". In broader terms, we propose that fundamental insights into cancer can be achieved by experimentation interacting with theoretical frameworks provided by computational and mathematical modeling.
Figures
References
-
- Moolgavkar SH, Luebeck EG. Multistage carcinogenesis and the incidence of human cancer. Genes Chromosomes Cancer. 2003;38(4):302–6. - PubMed
-
- Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1(2):157–62. - PubMed
-
- Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EY. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science. 1987;235(4794):1394–9. - PubMed
-
- Weinberg RA. Oncogenes and tumor suppressor genes. CA Cancer J Clin. 1994;44(3):160–70. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
