

Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria
- PMID: 18541666
- PMCID: PMC2519700
- DOI: 10.1128/MCB.00457-08
Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria
Abstract
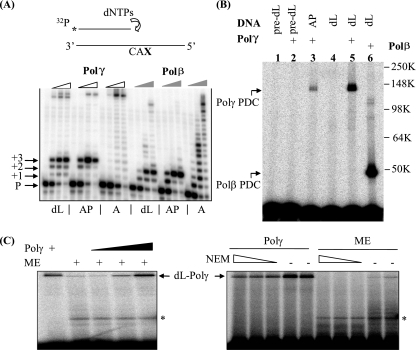
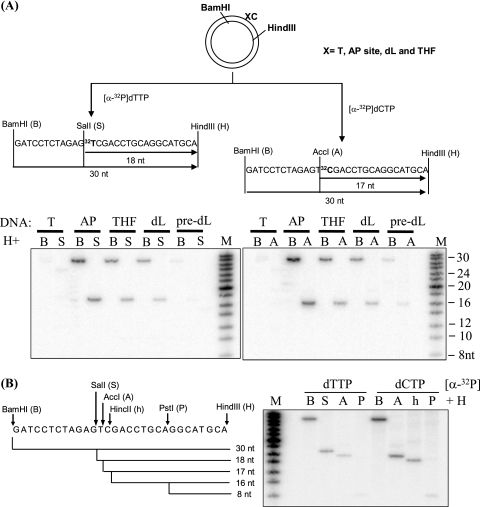
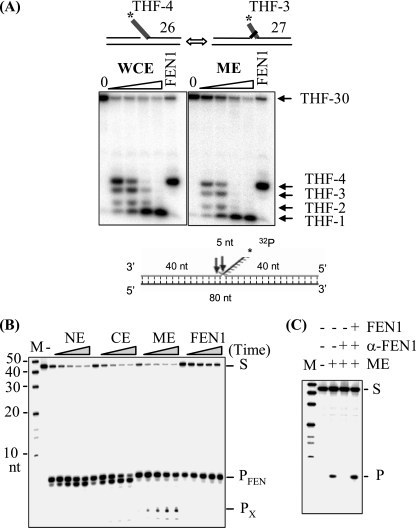
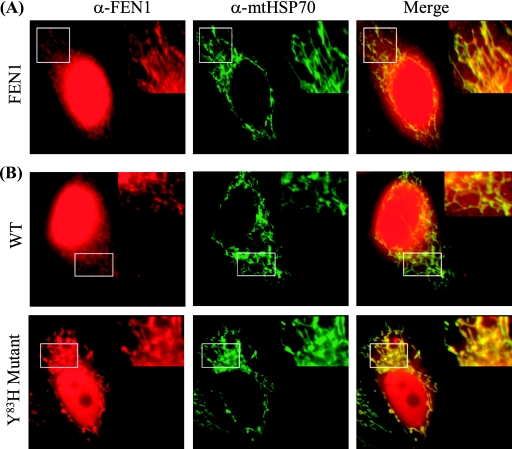
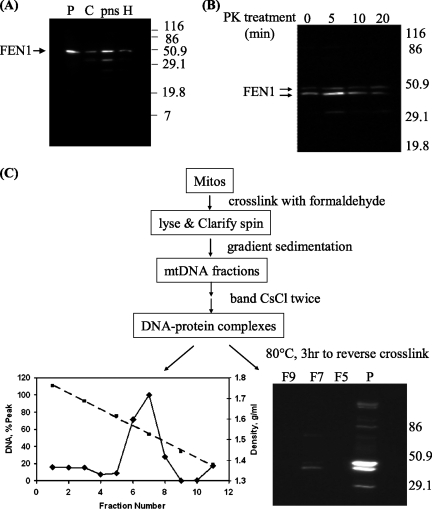
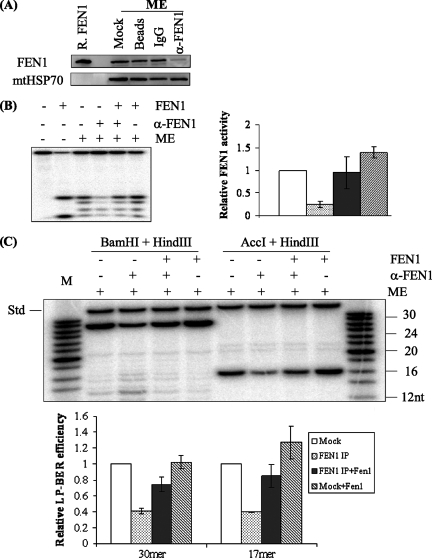
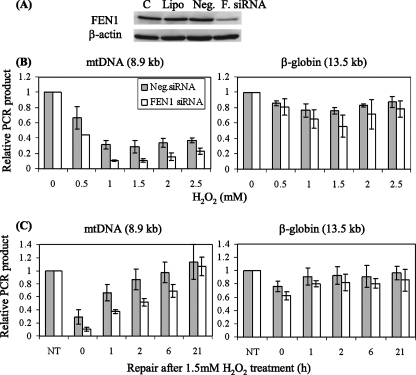
Repair of oxidative DNA damage in mitochondria was thought limited to short-patch base excision repair (SP-BER) replacing a single nucleotide. However, certain oxidative lesions cannot be processed by SP-BER. Here we report that 2-deoxyribonolactone (dL), a major type of oxidized abasic site, inhibits replication by mitochondrial DNA (mtDNA) polymerase gamma and interferes with SP-BER by covalently trapping polymerase gamma during attempted dL excision. However, repair of dL was detected in human mitochondrial extracts, and we show that this repair is via long-patch BER (LP-BER) dependent on flap endonuclease 1 (FEN1), not previously known to be present in mitochondria. FEN1 was retained in protease-treated mitochondria and detected in mitochondrial nucleoids that contain known mitochondrial replication and transcription proteins. Results of immunofluorescence and subcellular fractionation studies were also consistent with the presence of FEN1 in the mitochondria of intact cells. Immunodepletion experiments showed that the LP-BER activity of mitochondrial extracts was strongly diminished in parallel with the removal of FEN1, although some activity remained, suggesting the presence of an additional flap-removing enzyme. Biological evidence for a FEN1 role in repairing mitochondrial oxidative DNA damage was provided by RNA interference experiments, with the extent of damage greater and the recovery slower in FEN1-depleted cells than in control cells. The mitochondrial LP-BER pathway likely plays important roles in repairing dL lesions and other oxidative lesions and perhaps in normal mtDNA replication.
Figures
References
-
- Akbari, M., T. Visnes, H. E. Krokan, and M. Otterlei. 2008. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair (Amsterdam) 7605-616. - PubMed
-
- Bender, A., K. J. Krishnan, C. M. Morris, G. A. Taylor, A. K. Reeve, R. H. Perry, E. Jaros, J. S. Hersheson, J. Betts, T. Klopstock, R. W. Taylor, and D. M. Turnbull. 2006. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38515-517. - PubMed
-
- Bogenhagen, D. F., D. Rousseau, and S. Burke. 2008. The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 2833665-3675. - PubMed
-
- Bohr, V. A. 2002. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic. Biol. Med. 32804-812. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous