

Protein model refinement using an optimized physics-based all-atom force field
- PMID: 18550813
- PMCID: PMC2448826
- DOI: 10.1073/pnas.0800054105
Protein model refinement using an optimized physics-based all-atom force field
Abstract
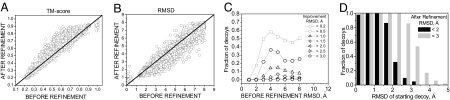
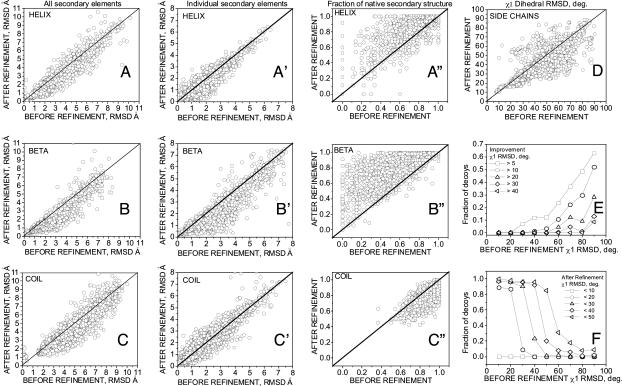
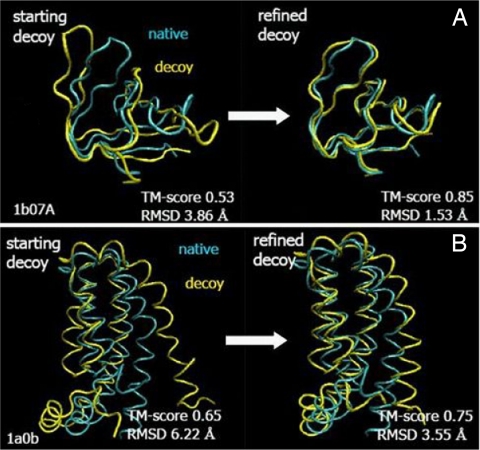
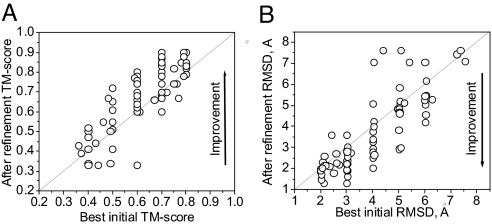
One of the greatest challenges in protein structure prediction is the refinement of low-resolution predicted models to high-resolution structures that are close to the native state. Although contemporary structure prediction methods can assemble the correct topology for a large fraction of protein domains, such approximate models are often not of the resolution required for many important applications, including studies of reaction mechanisms and virtual ligand screening. Thus, the development of a method that could bring those structures closer to the native state is of great importance. We recently optimized the relative weights of the components of the Amber ff03 potential on a large set of decoy structures to create a funnel-shaped energy landscape with the native structure at the global minimum. Such an energy function might be able to drive proteins toward their native structure. In this work, for a test set of 47 proteins, with 100 decoy structures per protein that have a range of structural similarities to the native state, we demonstrate that our optimized potential can drive protein models closer to their native structure. Comparing the lowest-energy structure from each trajectory with the starting decoy, structural improvement is seen for 70% of the models on average. The ability to do such systematic structural refinements by using a physics-based all-atom potential represents a promising approach to high-resolution structure prediction.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Zhang Y, Arakaki AK, Skolnick J. TASSER: An automated method for the prediction of protein tertiary structures in CASP6. Proteins. 2005;61(Suppl 7):91–98. - PubMed
-
- Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. - PubMed
-
- Fisher D. 3D-SHOTGUN: A novel, cooperative, fold-recognition meta-predictor. Proteins. 2003;51:434–441. - PubMed
-
- Kolinski A, Bujnicki J. Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models. Proteins. 2005;61:84–90. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
