

Phosphorylation of GSK-3beta by cGMP-dependent protein kinase II promotes hypertrophic differentiation of murine chondrocytes
- PMID: 18551195
- PMCID: PMC2423867
- DOI: 10.1172/JCI35243
Phosphorylation of GSK-3beta by cGMP-dependent protein kinase II promotes hypertrophic differentiation of murine chondrocytes
Erratum in
- J Clin Invest. 2008 Aug;118(8):2986
Abstract
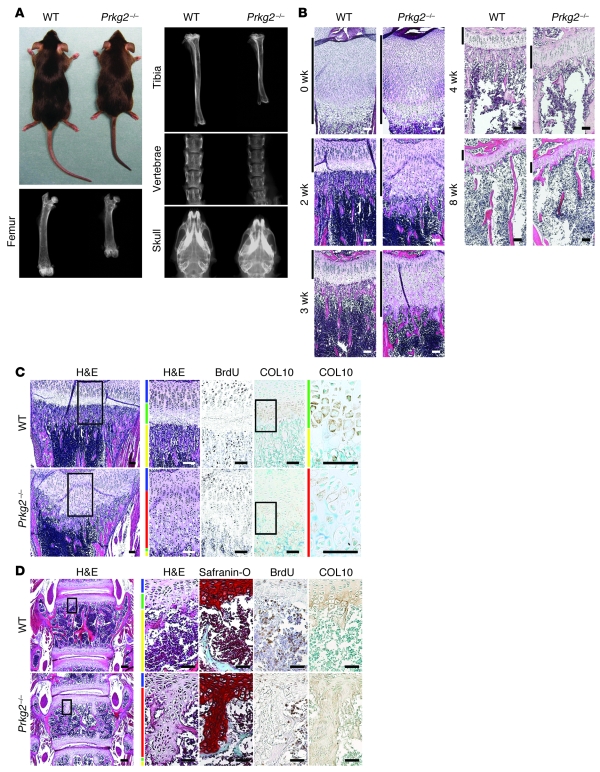
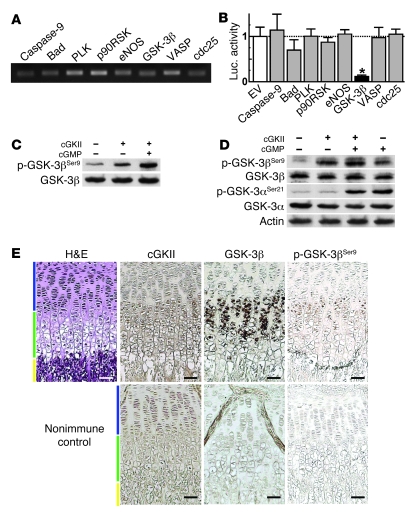
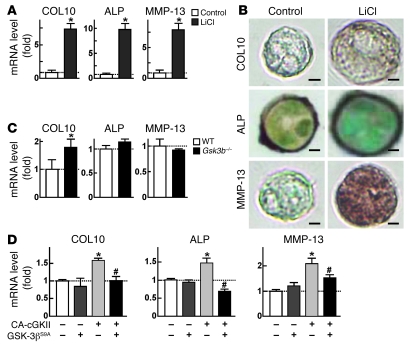
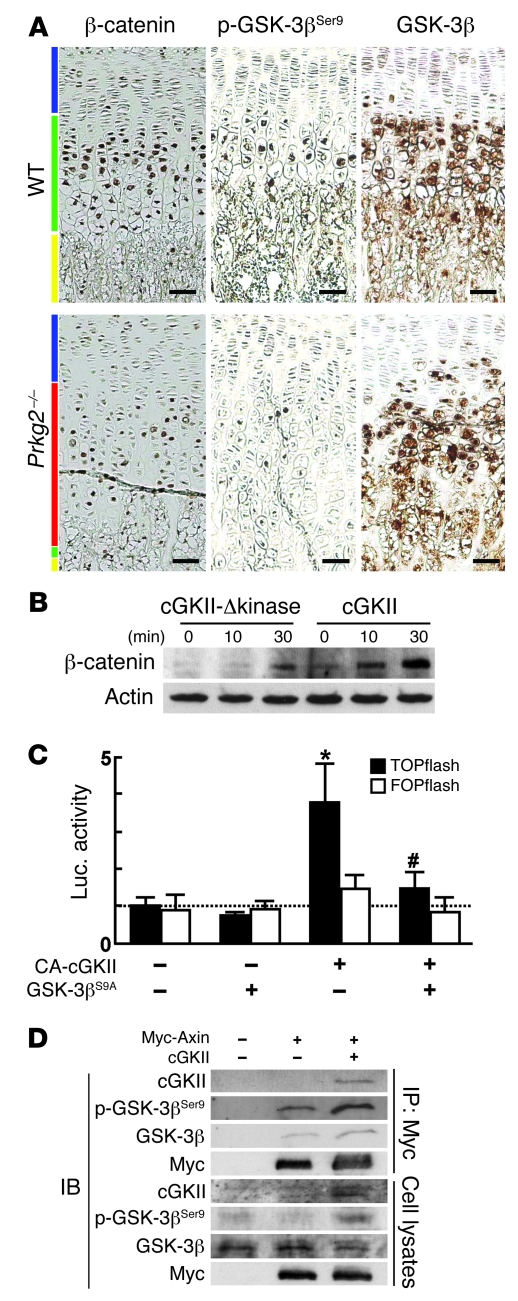
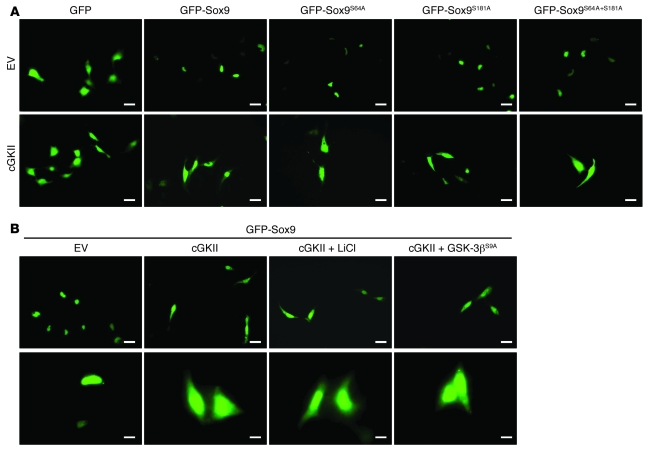
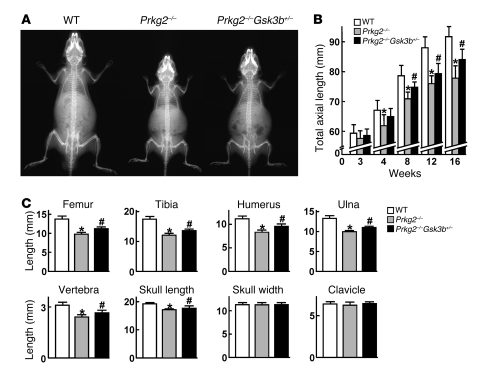
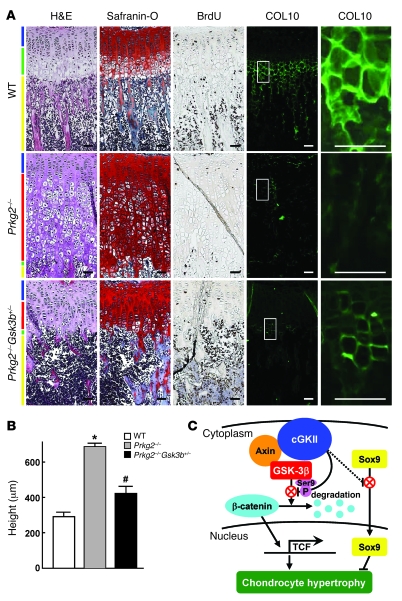
cGMP-dependent protein kinase II (cGKII; encoded by PRKG2) is a serine/threonine kinase that is critical for skeletal growth in mammals; in mice, cGKII deficiency results in dwarfism. Using radiographic analysis, we determined that this growth defect was a consequence of an elongated growth plate and impaired chondrocyte hypertrophy. To investigate the mechanism of cGKII-mediated chondrocyte hypertrophy, we performed a kinase substrate array and identified glycogen synthase kinase-3beta (GSK-3beta; encoded by Gsk3b) as a principal phosphorylation target of cGKII. In cultured mouse chondrocytes, phosphorylation-mediated inhibition of GSK-3beta was associated with enhanced hypertrophic differentiation. Furthermore, cGKII induction of chondrocyte hypertrophy was suppressed by cotransfection with a phosphorylation-deficient mutant of GSK-3beta. Analyses of mice with compound deficiencies in both protein kinases (Prkg2(-/-)Gsk3b(+/-)) demonstrated that the growth retardation and elongated growth plate associated with cGKII deficiency were partially rescued by haploinsufficiency of Gsk3b. We found that beta-catenin levels decreased in Prkg2(-/-) mice, while overexpression of cGKII increased the accumulation and transactivation function of beta-catenin in mouse chondroprogenitor ATDC5 cells. This effect was blocked by coexpression of phosphorylation-deficient GSK-3beta. These data indicate that hypertrophic differentiation of growth plate chondrocytes during skeletal growth is promoted by phosphorylation and inactivation of GSK-3beta by cGKII.
Figures
Similar articles
-
GSK-3α and GSK-3β proteins are involved in early stages of chondrocyte differentiation with functional redundancy through RelA protein phosphorylation.J Biol Chem. 2012 Aug 24;287(35):29227-36. doi: 10.1074/jbc.M112.372086. Epub 2012 Jul 3. J Biol Chem. 2012. PMID: 22761446 Free PMC article.
-
Cyclic GMP-dependent protein kinase II plays a critical role in C-type natriuretic peptide-mediated endochondral ossification.Endocrinology. 2002 Sep;143(9):3604-10. doi: 10.1210/en.2002-220307. Endocrinology. 2002. PMID: 12193576
-
cGMP-dependent protein kinase II determines β-catenin accumulation that is essential for uterine decidualization in mice.Am J Physiol Cell Physiol. 2019 Dec 1;317(6):C1115-C1127. doi: 10.1152/ajpcell.00208.2019. Epub 2019 Sep 11. Am J Physiol Cell Physiol. 2019. PMID: 31509448
-
Mutation in cGMP-dependent protein kinase II causes dwarfism in a rat mutant KMI through uncoupling of proliferation and differentiation of chondrocytes.J Bone Miner Metab. 2005;23(3):200-4. doi: 10.1007/s00774-004-0598-8. J Bone Miner Metab. 2005. PMID: 15838621 Review. No abstract available.
-
Glycogen synthase kinase 3: a key regulator of cellular fate.Cell Mol Life Sci. 2007 Aug;64(15):1930-44. doi: 10.1007/s00018-007-7045-7. Cell Mol Life Sci. 2007. PMID: 17530463 Free PMC article. Review.
Cited by
-
The increase in O-linked N-acetylglucosamine protein modification stimulates chondrogenic differentiation both in vitro and in vivo.J Biol Chem. 2012 Sep 28;287(40):33615-28. doi: 10.1074/jbc.M112.354241. Epub 2012 Aug 2. J Biol Chem. 2012. PMID: 22859309 Free PMC article.
-
GSK-3α and GSK-3β proteins are involved in early stages of chondrocyte differentiation with functional redundancy through RelA protein phosphorylation.J Biol Chem. 2012 Aug 24;287(35):29227-36. doi: 10.1074/jbc.M112.372086. Epub 2012 Jul 3. J Biol Chem. 2012. PMID: 22761446 Free PMC article.
-
Genetic deletion of Sphk2 confers protection against Pseudomonas aeruginosa mediated differential expression of genes related to virulent infection and inflammation in mouse lung.BMC Genomics. 2019 Dec 16;20(1):984. doi: 10.1186/s12864-019-6367-9. BMC Genomics. 2019. PMID: 31842752 Free PMC article.
-
Advances in Skeletal Dysplasia Genetics.Annu Rev Genomics Hum Genet. 2015;16:199-227. doi: 10.1146/annurev-genom-090314-045904. Epub 2015 Apr 22. Annu Rev Genomics Hum Genet. 2015. PMID: 25939055 Free PMC article. Review.
-
cGMP-Dependent Protein Kinase Inhibitors in Health and Disease.Pharmaceuticals (Basel). 2013 Feb 7;6(2):269-86. doi: 10.3390/ph6020269. Pharmaceuticals (Basel). 2013. PMID: 24275951 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
