

Combinatorial patterns of histone acetylations and methylations in the human genome
- PMID: 18552846
- PMCID: PMC2769248
- DOI: 10.1038/ng.154
Combinatorial patterns of histone acetylations and methylations in the human genome
Abstract
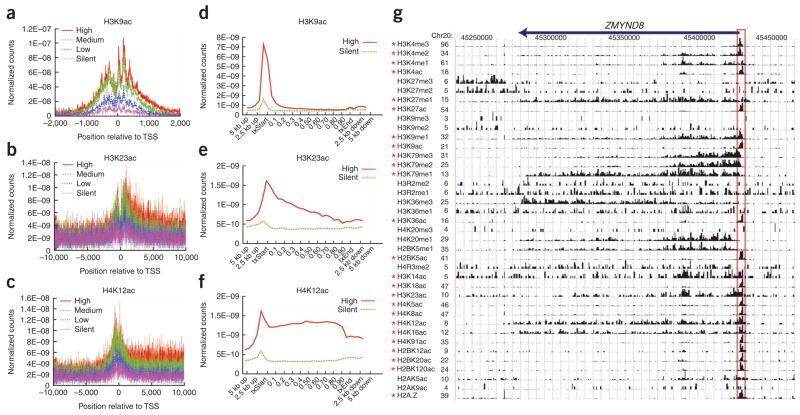
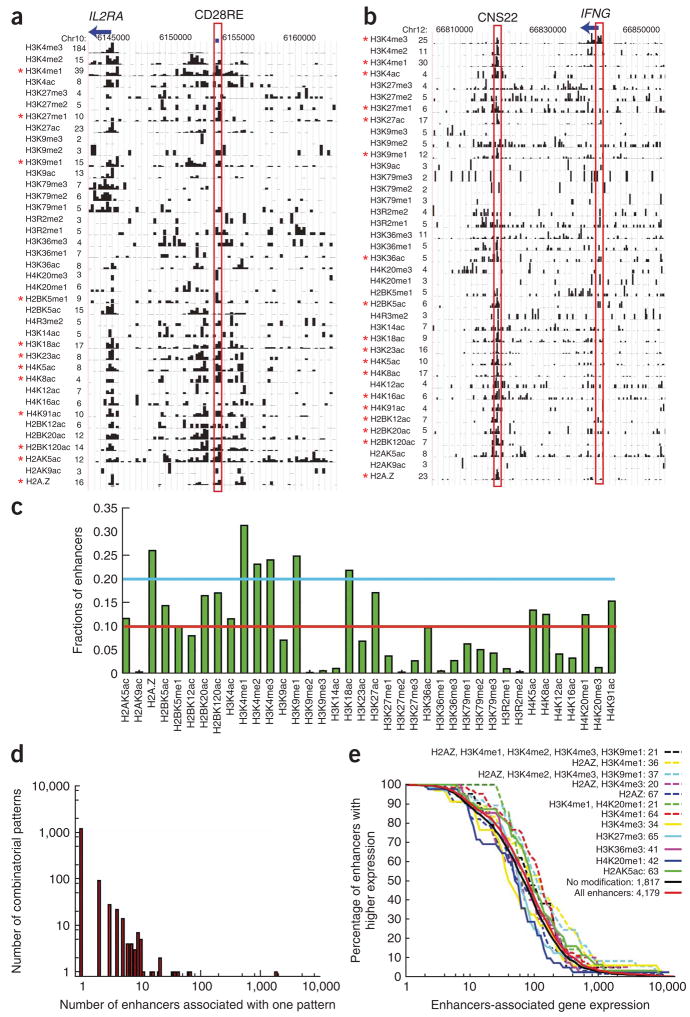
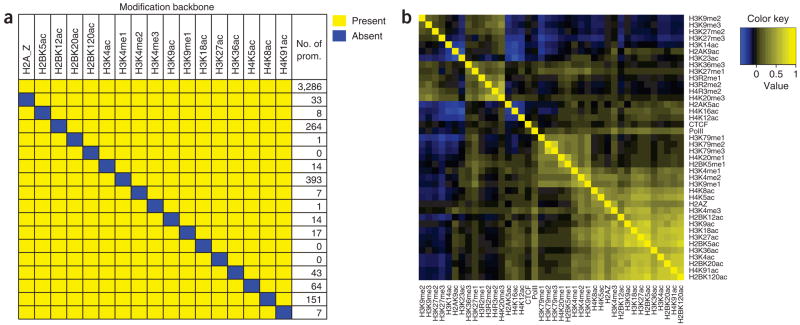
Histones are characterized by numerous posttranslational modifications that influence gene transcription. However, because of the lack of global distribution data in higher eukaryotic systems, the extent to which gene-specific combinatorial patterns of histone modifications exist remains to be determined. Here, we report the patterns derived from the analysis of 39 histone modifications in human CD4(+) T cells. Our data indicate that a large number of patterns are associated with promoters and enhancers. In particular, we identify a common modification module consisting of 17 modifications detected at 3,286 promoters. These modifications tend to colocalize in the genome and correlate with each other at an individual nucleosome level. Genes associated with this module tend to have higher expression, and addition of more modifications to this module is associated with further increased expression. Our data suggest that these histone modifications may act cooperatively to prepare chromatin for transcriptional activation.
Figures


References
-
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. - PubMed
-
- Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. - PubMed
-
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. - PubMed
-
- Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–2360. - PubMed
-
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
