

Diagnostic and mutational spectrum of progressive osseous heteroplasia (POH) and other forms of GNAS-based heterotopic ossification
- PMID: 18553568
- PMCID: PMC2564798
- DOI: 10.1002/ajmg.a.32346
Diagnostic and mutational spectrum of progressive osseous heteroplasia (POH) and other forms of GNAS-based heterotopic ossification
Abstract
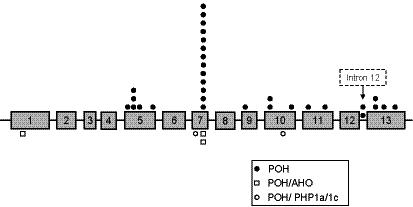
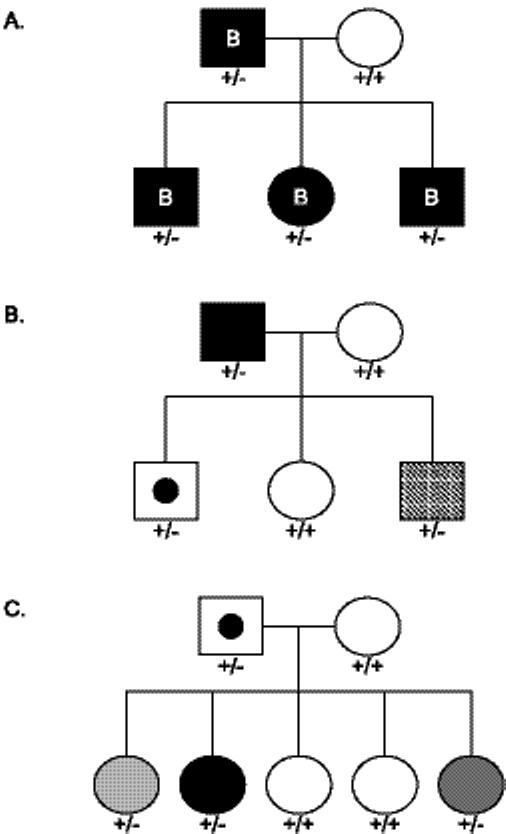
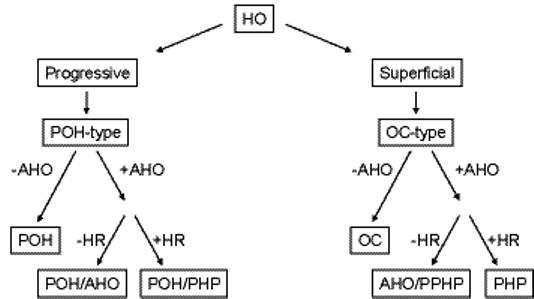
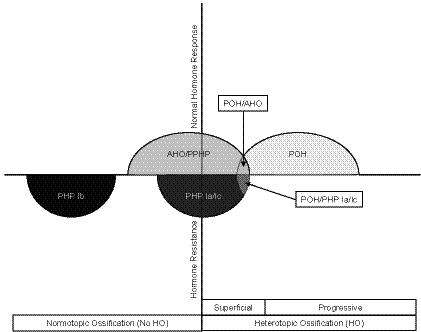
Progressive osseous heteroplasia (POH) is a rare, disabling disease of heterotopic ossification (HO) that progresses from skin and subcutaneous tissues into deep skeletal muscle. POH occurs in the absence of multiple developmental features of Albright hereditary osteodystrophy (AHO) or hormone resistance, clinical manifestations that are also associated with GNAS inactivation. However, occasional patients with AHO and pseudohypoparathyroidism 1a/c (PHP1a/c; AHO features plus hormone resistance) have also been described who have progressive HO. This study was undertaken to define the diagnostic and mutational spectrum of POH and progressive disorders of HO, and to distinguish them from related disorders in which HO remains confined to the skin and subcutaneous tissues. We reviewed the charts of 111 individuals who had cutaneous and subcutaneous ossification. All patients were assessed for eight characteristics: age of onset of HO, presence and location of HO, depth of HO, type of HO, progression of HO, features of AHO, PTH resistance, and GNAS mutation analysis. We found, based on clinical criteria, that POH and progressive HO syndromes are at the severe end of a phenotypic spectrum of GNAS-inactivating conditions associated with extra-skeletal ossification. While most individuals with superficial or progressive ossification had mutations in GNAS, there were no specific genotype-phenotype correlations that distinguished the more progressive forms of HO (e.g., POH) from the non-progressive forms (osteoma cutis, AHO, and PHP1a/c).
2008 Wiley-Liss, Inc.
Figures
References
-
- Ahmed SF, Dixon PH, Bonthron DT, Stirling HF, Barr DG, Kelnar CJ, Thakker RV. GNAS1 mutational analysis in pseudohypoparathyroidism. Clin Endocrinol (Oxf) 1998;49:525–531. - PubMed
-
- Ahrens W, Hiort O, Staedt P, Kirschner T, Marschke C, Kruse K. Analysis of the GNAS1 gene in Albright's hereditary osteodystrophy. J Clin Endocrinol Metab. 2001;86:4630–4634. - PubMed
-
- Aldred MA, Trembath RC. Activating and inactivating mutations in the human GNAS1 gene. Hum Mutat. 2000;16:183–189. - PubMed
-
- Bastepe M, Frohlich LF, Hendy GN, Indridason OS, Josse RG, Koshiyama H, Korkko J, Nakamoto JM, Rosenbloom AL, Slyper AH, Sugimoto T, Tsatsoulis A, Crawford JD, Juppner H. Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J Clin Invest. 2003;112:1255–1263. - PMC - PubMed
-
- Bastepe M, Frohlich LF, Linglart A, Abu-Zahra HS, Tojo K, Ward LM, Juppner H. Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypoparathyroidism type Ib. Nat Genet. 2005a;37:25–27. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
