

Improving the prediction accuracy in classification using the combined data sets by ranks of gene expressions
- PMID: 18554423
- PMCID: PMC2442106
- DOI: 10.1186/1471-2105-9-283
Improving the prediction accuracy in classification using the combined data sets by ranks of gene expressions
Abstract
Background: The information from different data sets experimented under different conditions may be inconsistent even though they are performed with the same research objectives. More than that, even when the data sets were generated from the same platform, the data agreement may be affected by the technical variation among the laboratories. In this case, it is necessary to use the combined data set after adjusting the differences between such data sets, for detecting the more reliable information.
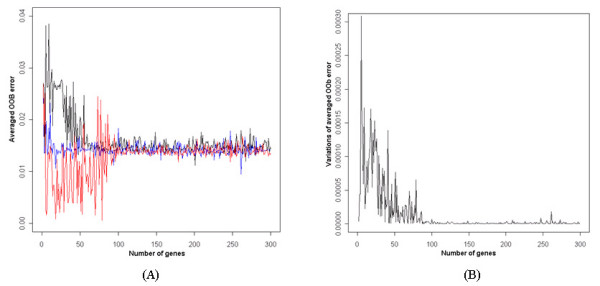
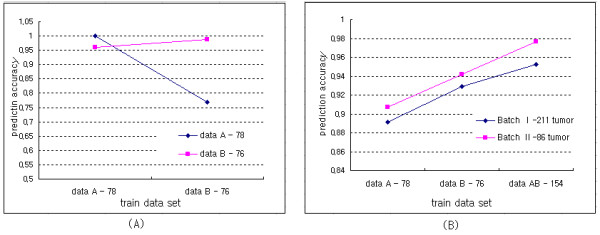
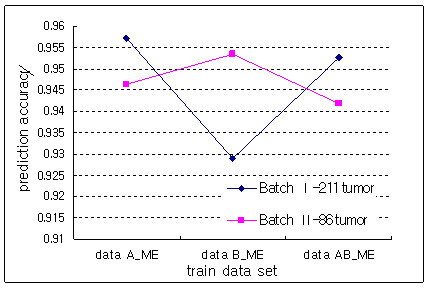
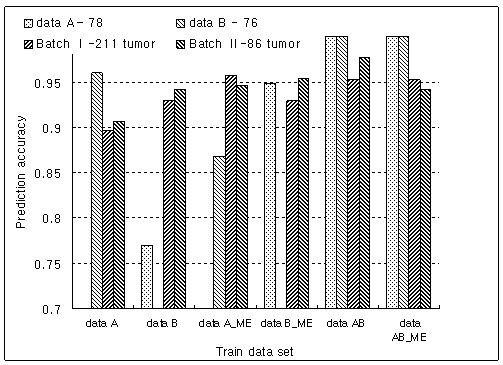
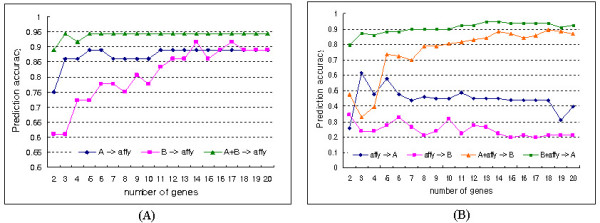
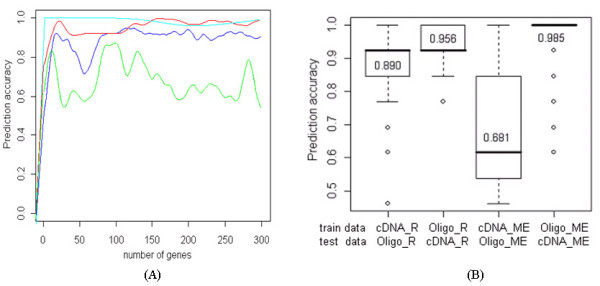
Results: The proposed method combines data sets posterior to the discretization of data sets based on the ranks of the gene expression ratios, and the statistical method is applied to the combined data set for predictive gene selection. The efficiency of the proposed method was evaluated using five colon cancer related data sets, which were experimented using cDNA microarrays with different RNA sources, and one experiment utilized oligonucleotide arrays. NCI-60 cell lines data sets were used, which were performed with two different platforms of cDNA microarrays and Affymetrix HU6800 oligonucleotide arrays. The combined data set by the proposed method predicted the test data sets more accurately than the separated data sets did. The biological significant genes were detected from the combined data set, which were missed on the separated data sets.
Conclusion: By transforming gene expressions using ranks, the proposed method is not influenced by systematic bias among chips and normalization method. The method may be especially more useful to find predictive genes from data sets which have different scale in gene expressions.
Figures
Similar articles
-
Methods for evaluating gene expression from Affymetrix microarray datasets.BMC Bioinformatics. 2008 Jun 17;9:284. doi: 10.1186/1471-2105-9-284. BMC Bioinformatics. 2008. PMID: 18559105 Free PMC article.
-
Novel and simple transformation algorithm for combining microarray data sets.BMC Bioinformatics. 2007 Jun 25;8:218. doi: 10.1186/1471-2105-8-218. BMC Bioinformatics. 2007. PMID: 17588268 Free PMC article.
-
Improving gene set analysis of microarray data by SAM-GS.BMC Bioinformatics. 2007 Jul 5;8:242. doi: 10.1186/1471-2105-8-242. BMC Bioinformatics. 2007. PMID: 17612399 Free PMC article.
-
Redefinition of Affymetrix probe sets by sequence overlap with cDNA microarray probes reduces cross-platform inconsistencies in cancer-associated gene expression measurements.BMC Bioinformatics. 2005 Apr 25;6:107. doi: 10.1186/1471-2105-6-107. BMC Bioinformatics. 2005. PMID: 15850491 Free PMC article.
-
Towards knowledge-based gene expression data mining.J Biomed Inform. 2007 Dec;40(6):787-802. doi: 10.1016/j.jbi.2007.06.005. Epub 2007 Jun 21. J Biomed Inform. 2007. PMID: 17683991 Review.
Cited by
-
A method for detecting significant genomic regions associated with oral squamous cell carcinoma using aCGH.Med Biol Eng Comput. 2010 May;48(5):459-68. doi: 10.1007/s11517-010-0595-0. Epub 2010 Mar 20. Med Biol Eng Comput. 2010. PMID: 20306232
-
Apontic directly activates hedgehog and cyclin E for proper organ growth and patterning.Sci Rep. 2017 Sep 29;7(1):12470. doi: 10.1038/s41598-017-12766-w. Sci Rep. 2017. PMID: 28963499 Free PMC article.
-
Possibility of the use of public microarray database for identifying significant genes associated with oral squamous cell carcinoma.Genomics Inform. 2012 Mar;10(1):23-32. doi: 10.5808/GI.2012.10.1.23. Epub 2012 Mar 31. Genomics Inform. 2012. PMID: 23105925 Free PMC article.
-
Conserved expression patterns predict microRNA targets.PLoS Comput Biol. 2009 Sep;5(9):e1000513. doi: 10.1371/journal.pcbi.1000513. Epub 2009 Sep 25. PLoS Comput Biol. 2009. PMID: 19779543 Free PMC article.
-
Development of novel predictive miRNA/target gene pathways for colorectal cancer distance metastasis to the liver using a bioinformatic approach.PLoS One. 2019 Feb 26;14(2):e0211968. doi: 10.1371/journal.pone.0211968. eCollection 2019. PLoS One. 2019. PMID: 30807603 Free PMC article.
References
-
- Hedges LV, Olkin I. Statistical Methods for Meta-Analysis. Orlando: Academic Press; 1985.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
