

Extracting sequence features to predict protein-DNA interactions: a comparative study
- PMID: 18556756
- PMCID: PMC2475627
- DOI: 10.1093/nar/gkn361
Extracting sequence features to predict protein-DNA interactions: a comparative study
Abstract
Predicting how and where proteins, especially transcription factors (TFs), interact with DNA is an important problem in biology. We present here a systematic study of predictive modeling approaches to the TF-DNA binding problem, which have been frequently shown to be more efficient than those methods only based on position-specific weight matrices (PWMs). In these approaches, a statistical relationship between genomic sequences and gene expression or ChIP-binding intensities is inferred through a regression framework; and influential sequence features are identified by variable selection. We examine a few state-of-the-art learning methods including stepwise linear regression, multivariate adaptive regression splines, neural networks, support vector machines, boosting and Bayesian additive regression trees (BART). These methods are applied to both simulated datasets and two whole-genome ChIP-chip datasets on the TFs Oct4 and Sox2, respectively, in human embryonic stem cells. We find that, with proper learning methods, predictive modeling approaches can significantly improve the predictive power and identify more biologically interesting features, such as TF-TF interactions, than the PWM approach. In particular, BART and boosting show the best and the most robust overall performance among all the methods.
Figures
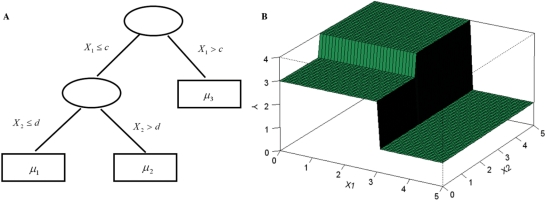
 ]. (B) The piece-wise constant function defined by the regression tree with c = 3, d = 2 and
]. (B) The piece-wise constant function defined by the regression tree with c = 3, d = 2 and  .
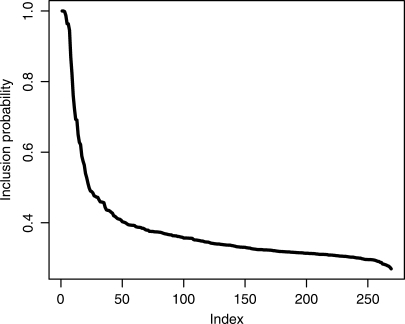
.
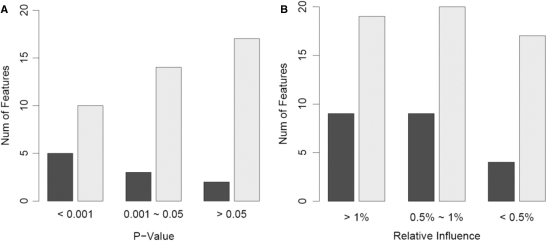
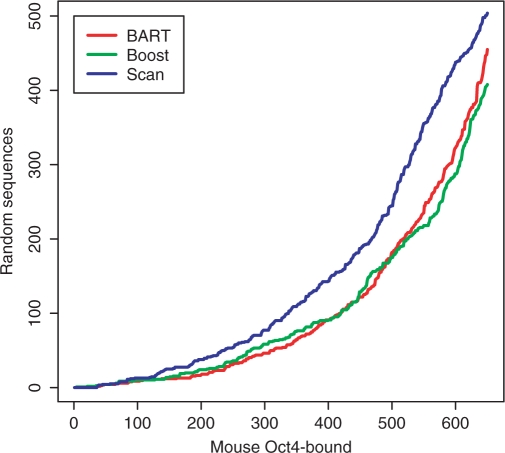
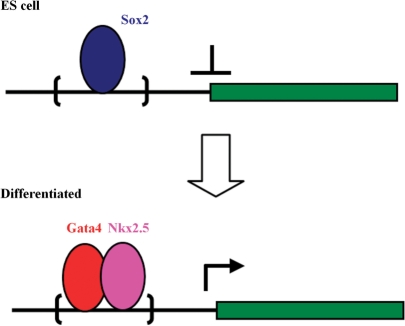
References
-
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wooton JC. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. - PubMed
-
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. - PubMed
-
- Liu X, Brutlag DL, Liu JS. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac. Symp. Biocomput. 2001;6:127–138. - PubMed
-
- Roth FR, Hughes JD, Estep PE, Church GM. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole genome mRNA quantization. Nat. Biotechnol. 1998;16:939–945. - PubMed
