

Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease
- PMID: 18558632
- PMCID: PMC2733806
- DOI: 10.1093/hmg/ddn175
Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease
Abstract
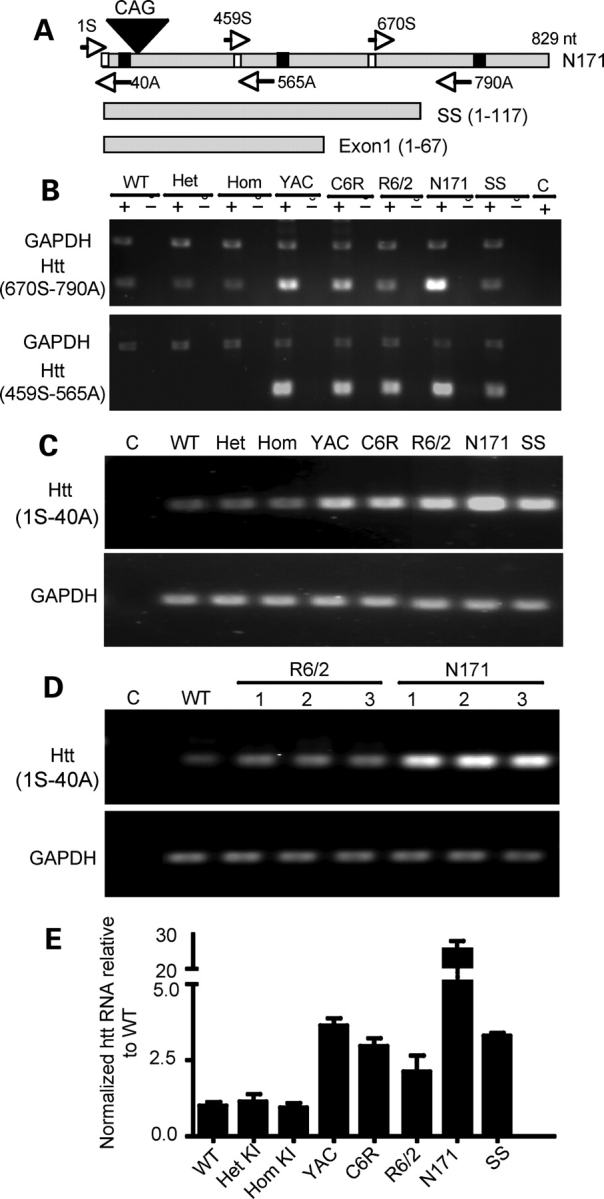
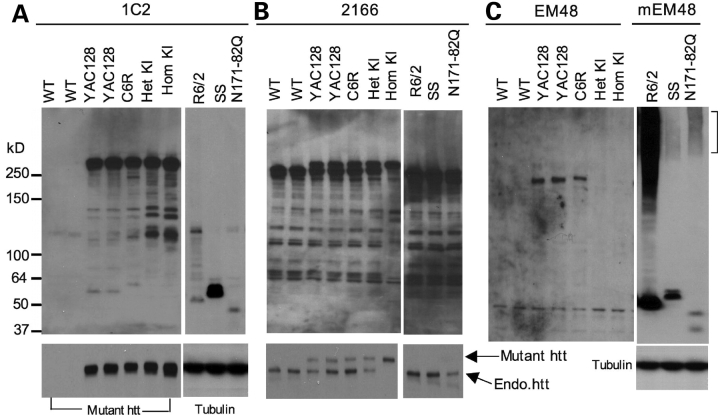
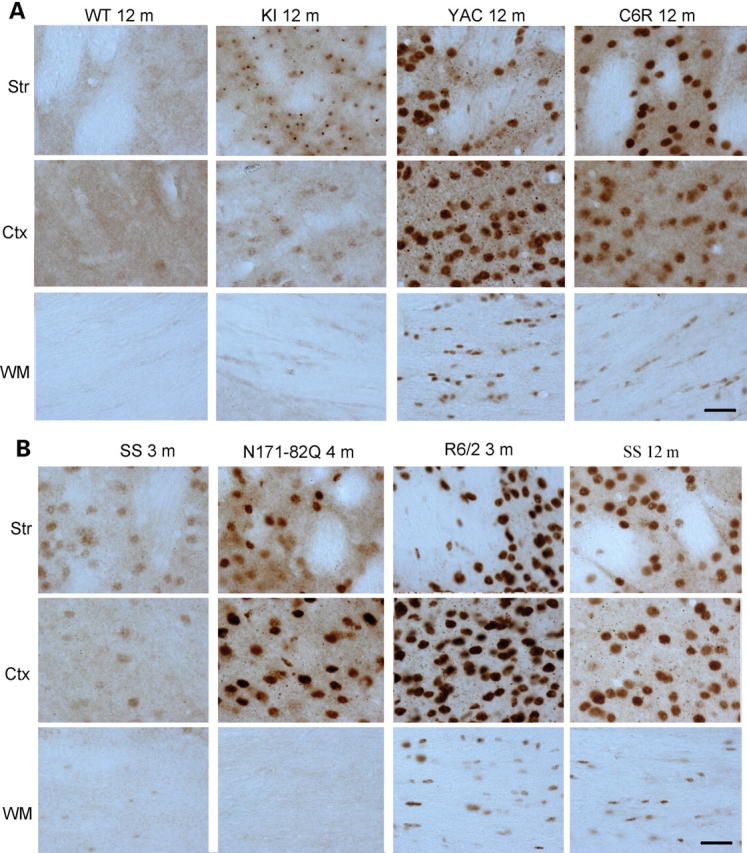
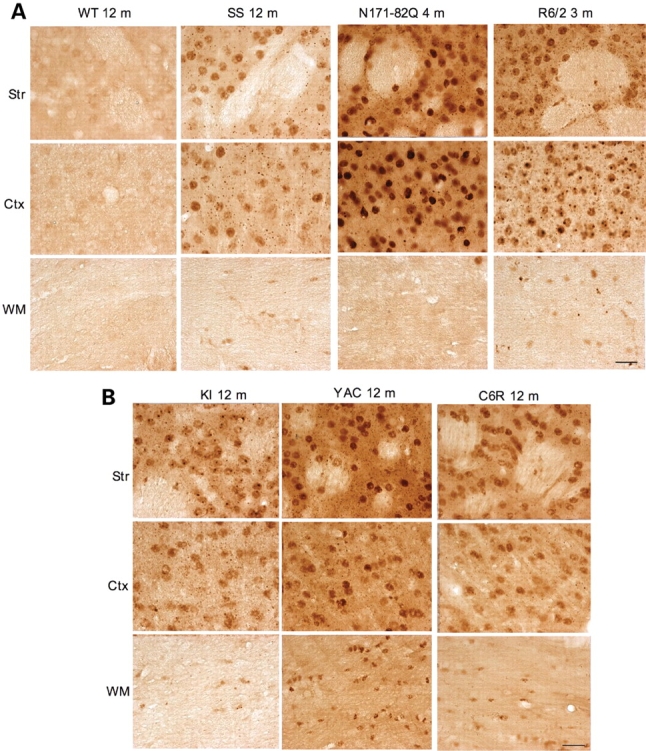
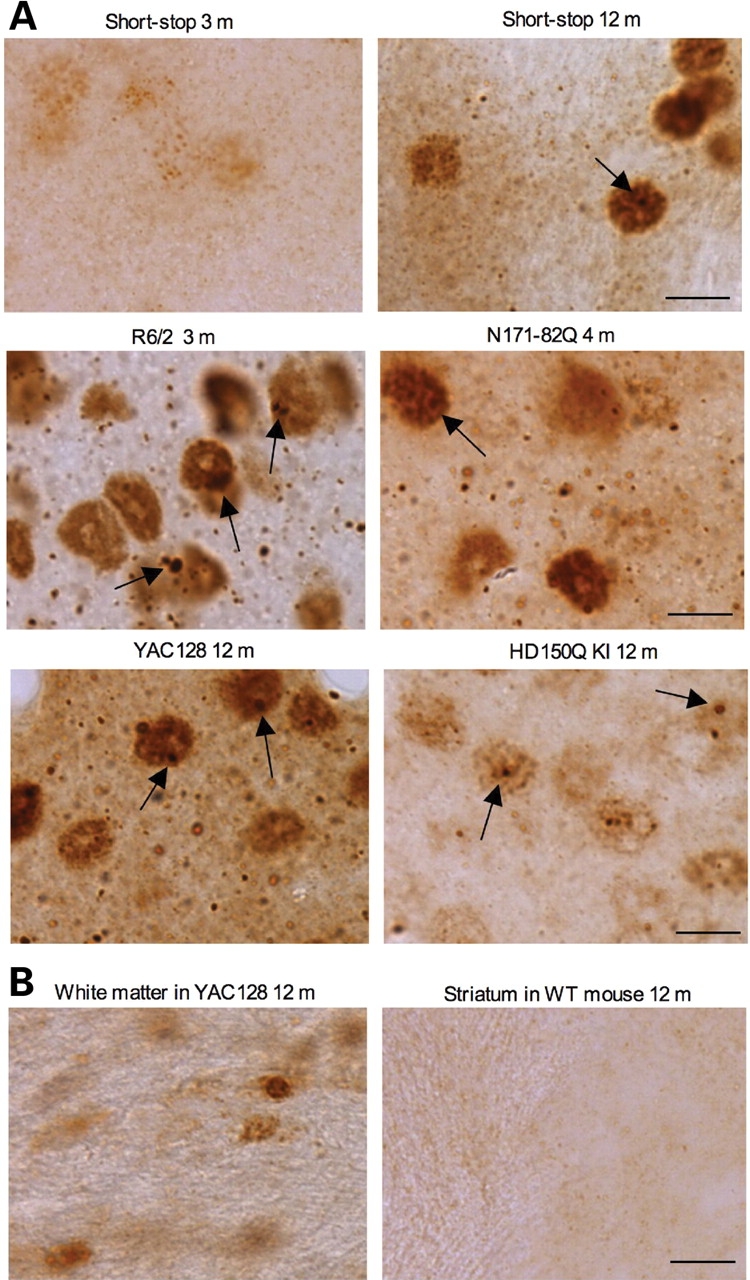
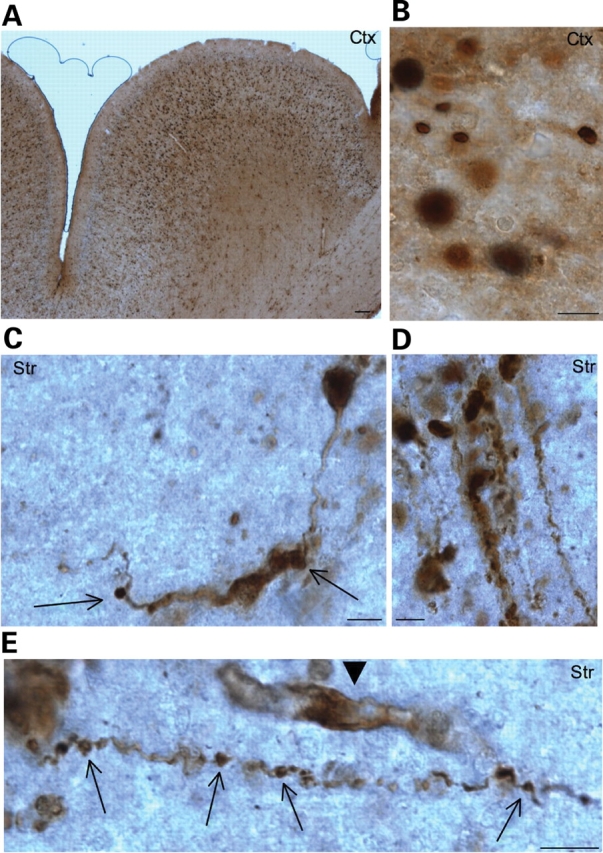
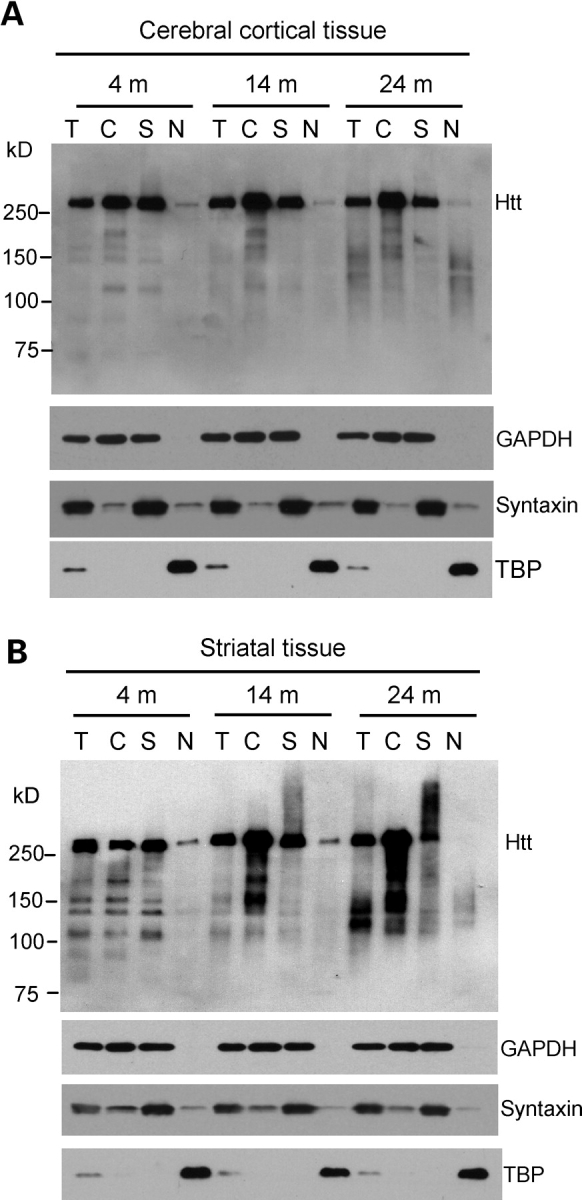
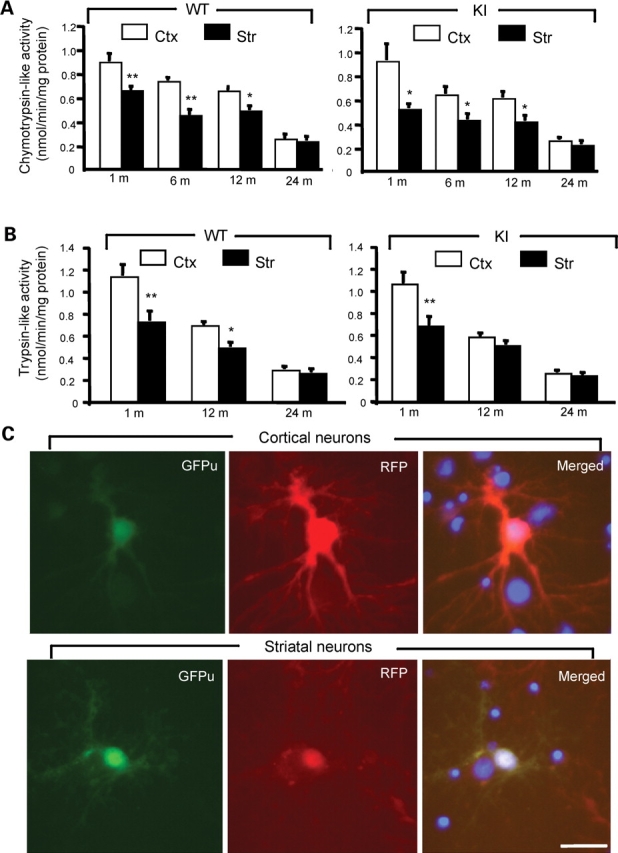
A number of mouse models expressing mutant huntingtin (htt) with an expanded polyglutamine (polyQ) domain are useful for studying the pathogenesis of Huntington's disease (HD) and identifying appropriate therapies. However, these models exhibit neurological phenotypes that differ in their severity and nature. Understanding how transgenic htt leads to variable neuropathology in animal models would shed light on the pathogenesis of HD and help us to choose HD models for investigation. By comparing the expression of mutant htt at the transcriptional and protein levels in transgenic mice expressing N-terminal or full-length mutant htt, we found that the accumulation and aggregation of mutant htt in the brain is determined by htt context. HD mouse models demonstrating more severe phenotypes show earlier accumulation of N-terminal mutant htt fragments, which leads to the formation of htt aggregates that are primarily present in neuronal nuclei and processes, as well as glial cells. Similarly, transgenic monkeys expressing exon-1 htt with a 147-glutamine repeat (147Q) died early and showed abundant neuropil aggregates in swelling neuronal processes. Fractionation of HD150Q knock-in mice brains revealed an age-dependent accumulation of N-terminal mutant htt fragments in the nucleus and synaptosomes, and this accumulation was most pronounced in the striatum due to decreased proteasomal activity. Our findings suggest that the neuropathological phenotypes of HD stem largely from the accumulation of N-terminal mutant htt fragments and that this accumulation is determined by htt context and cell-type-dependent clearance of mutant htt.
Figures
References
-
- Gusella J.F., Macdonald M.E. Huntington's disease: seeing the pathogenic process through a genetic lens. Trends Biochem. Sci. 2006;31:533–540. - PubMed
-
- Li S.H., Li X.J. Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 2004;20:146–154. - PubMed
-
- Vonsattel J.P., Myers R.H., Stevens T.J., Ferrante R.J., Bird E.D., Richardson E.P., Jr Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 1985;44:559–577. - PubMed
-
- Martin J.B., Gusella J.F. Huntington's disease. Pathogenesis and management. N. Engl. J. Med. 1986;315:1267–1276. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
