

PTHR1 mutations associated with Ollier disease result in receptor loss of function
- PMID: 18559376
- PMCID: PMC2722890
- DOI: 10.1093/hmg/ddn176
PTHR1 mutations associated with Ollier disease result in receptor loss of function
Abstract
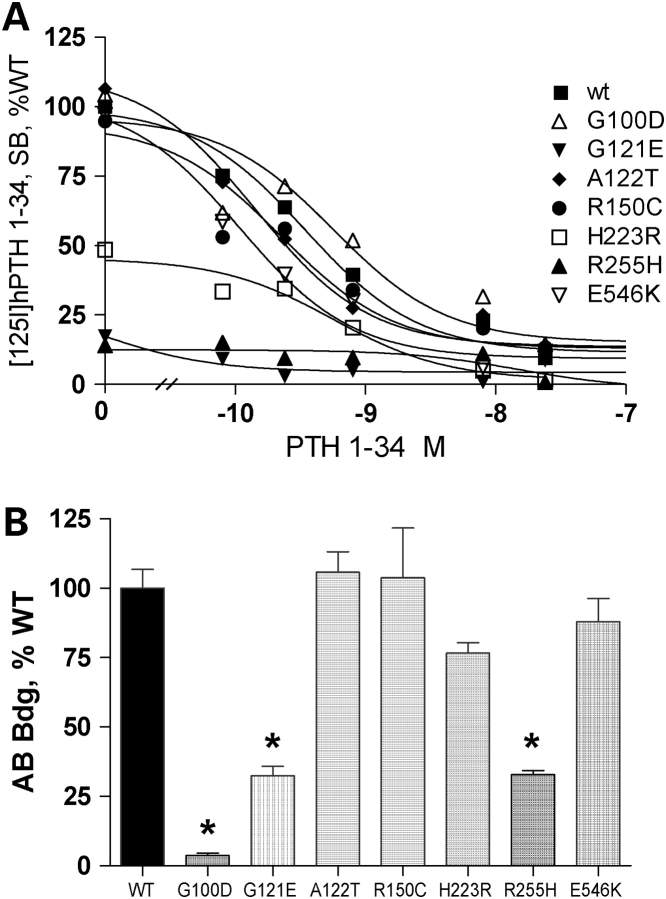
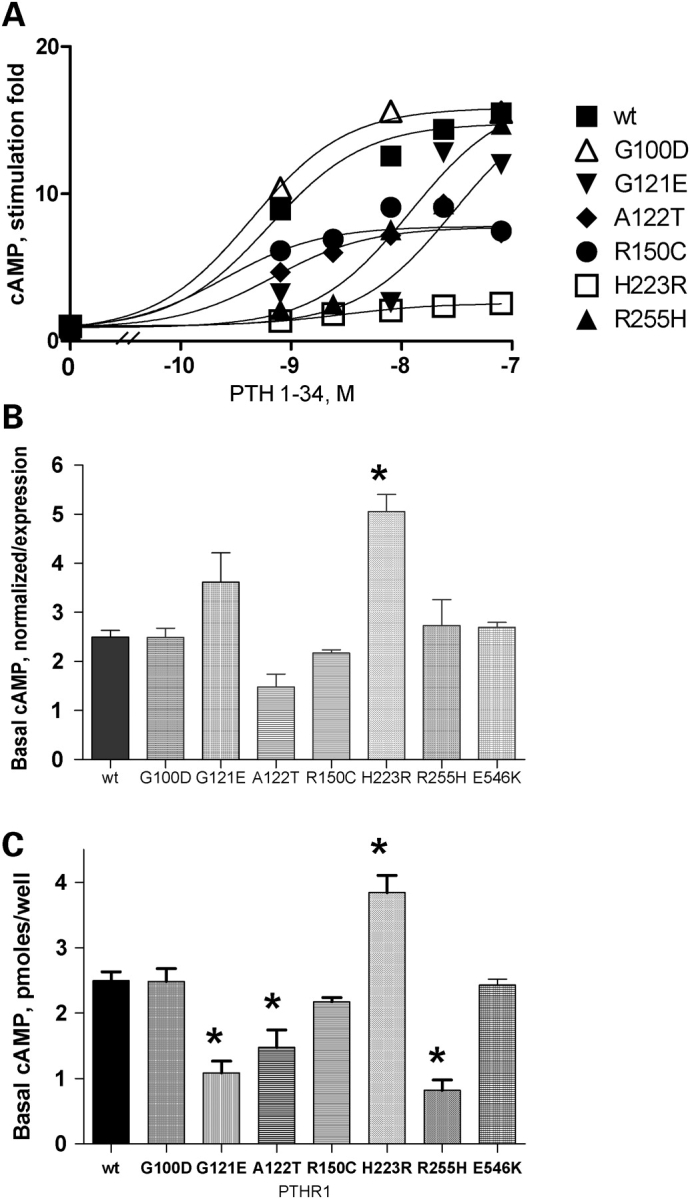
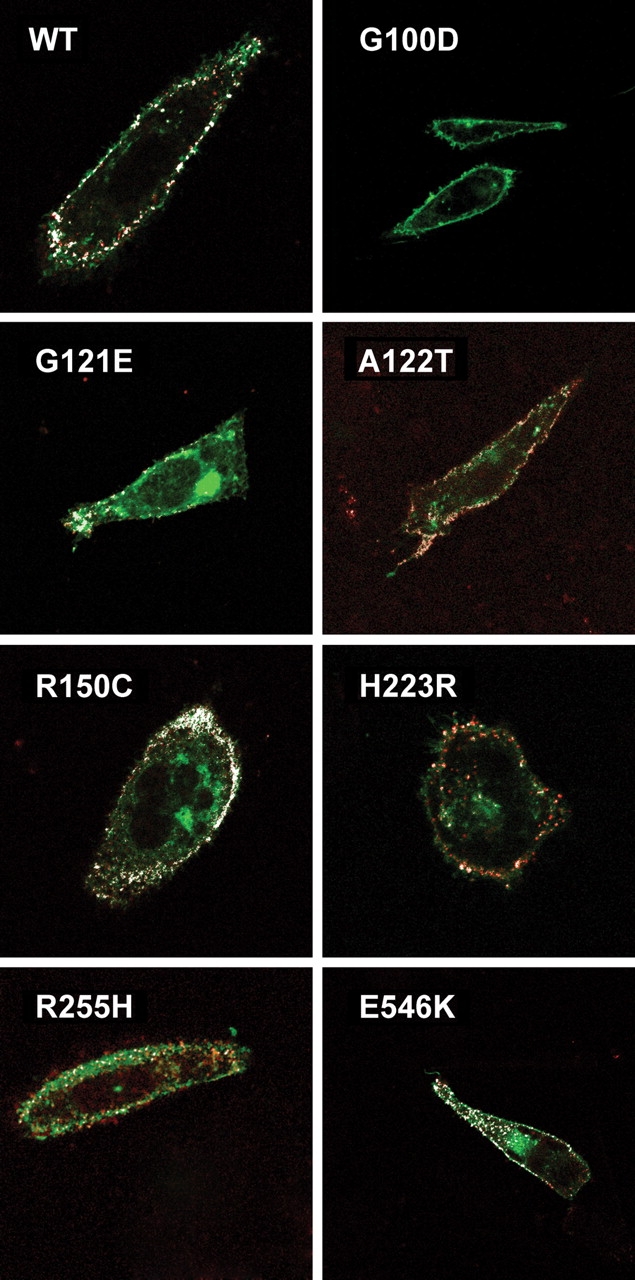
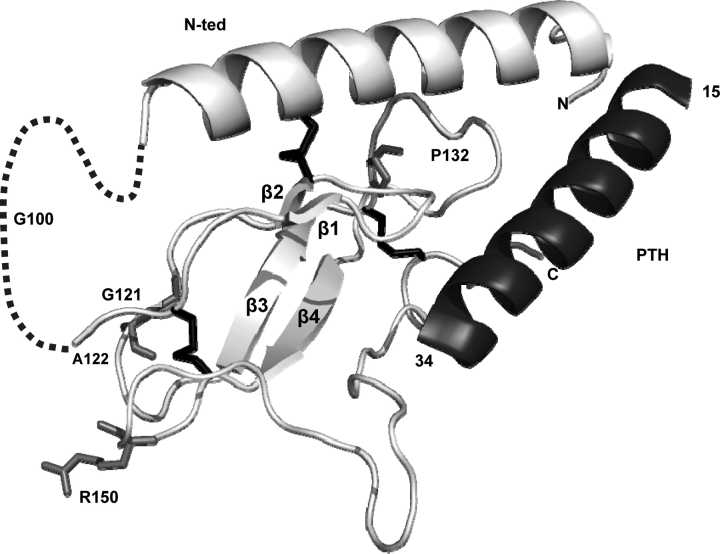
PTHR1-signaling pathway is critical for the regulation of endochondral ossification. Thus, abnormalities in genes belonging to this pathway could potentially participate in the pathogenesis of Ollier disease/Maffucci syndrome, two developmental disorders defined by the presence of multiple enchondromas. In agreement, a functionally deleterious mutation in PTHR1 (p.R150C) was identified in enchondromas from two of six unrelated patients with enchondromatosis. However, neither the p.R150C mutation (26 tumors) nor any other mutation in the PTHR1 gene (11 patients) could be identified in another study. To further define the role of PTHR1-signaling pathway in Ollier disease and Maffucci syndrome, we analyzed the coding sequences of four genes (PTHR1, IHH, PTHrP and GNAS1) in leucocyte and/or tumor DNA from 61 and 23 patients affected with Ollier disease or Maffucci syndrome, respectively. We identified three previously undescribed missense mutations in PTHR1 in patients with Ollier disease at the heterozygous state. Two mutations (p.G121E, p.A122T) were present only in enchondromas, and one (p.R255H) in both enchondroma and leukocyte DNA. Assessment of receptor function demonstrated that these three mutations impair PTHR1 function by reducing either the affinity of the receptor for PTH or the receptor expression at the cell surface. These mutations were not found in DNA from 222 controls. Including our data, PTHR1 functionally deleterious mutations have now been identified in five out 31 enchondromas from Ollier patients. These findings provide further support for the idea that heterozygous mutations in PTHR1 that impair receptor function participate in the pathogenesis of Ollier disease in some patients.
Figures
References
-
- Fletcher C.D.M., Unni K., Mertens F., editors. World Health Organization Classification of Tumors. Pathology and Genetics. Tumors of Soft Tissue and Bone. Lyon: IARCPress; 2002.
-
- Maroteaux P., Le Merrer M. Les maladies osseuses de l’enfant. 4ème ed. Paris: Médecine-Sciences, Flammarion; 2002.
-
- Unni K.K. Cartilaginous lesions of bone. J. Orthop. Sci. 2001;6:457–472. - PubMed
-
- Whyte M. Acquired Disorders of Cartilage and Bone. 5th edn. Washington, DC: American Society for Bone and Mineral Research; 2003.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
