

Allostery and cooperativity revisited
- PMID: 18560010
- PMCID: PMC2492820
- DOI: 10.1110/ps.03259908
Allostery and cooperativity revisited
Abstract
Although phenomenlogical models that account for cooperativity in allosteric systems date back to the early and mid-60's (e.g., the KNF and MWC models), there is resurgent interest in the topic due to the recent experimental and computational studies that attempted to reveal, at an atomistic level, how allostery actually works. In this review, using systems for which atomistic simulations have been carried out in our groups as examples, we describe the current understanding of allostery, how the mechanisms go beyond the classical MWC/Pauling-KNF descriptions, and point out that the "new view" of allostery, emphasizing "population shifts," is, in fact, an "old view." The presentation offers not only an up-to-date description of allostery from a theoretical/computational perspective, but also helps to resolve several outstanding issues concerning allostery.
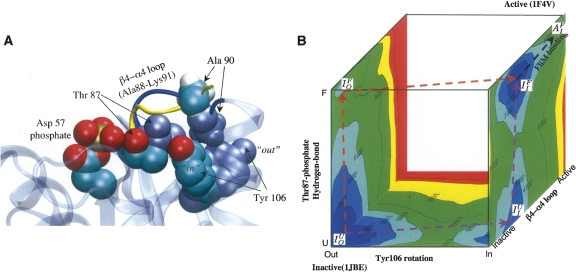
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
