

Knockdown of cytosolic glutaredoxin 1 leads to loss of mitochondrial membrane potential: implication in neurodegenerative diseases
- PMID: 18560520
- PMCID: PMC2426930
- DOI: 10.1371/journal.pone.0002459
Knockdown of cytosolic glutaredoxin 1 leads to loss of mitochondrial membrane potential: implication in neurodegenerative diseases
Abstract
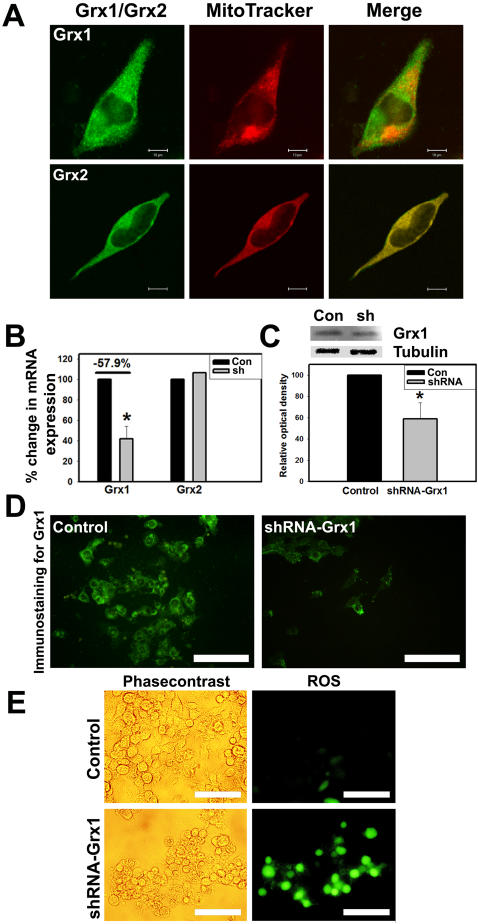
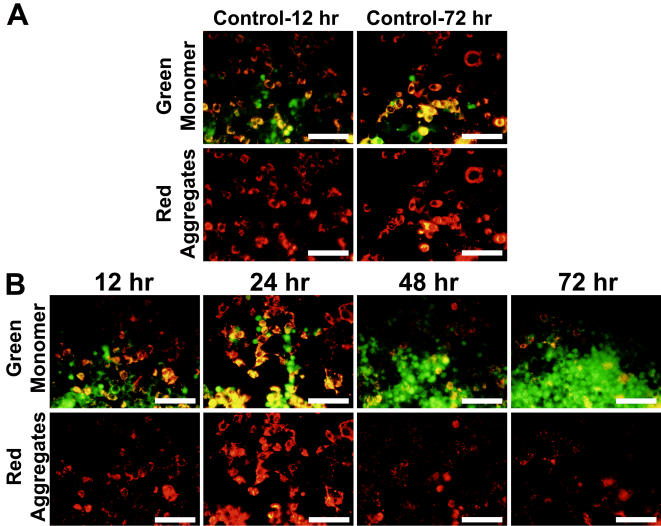
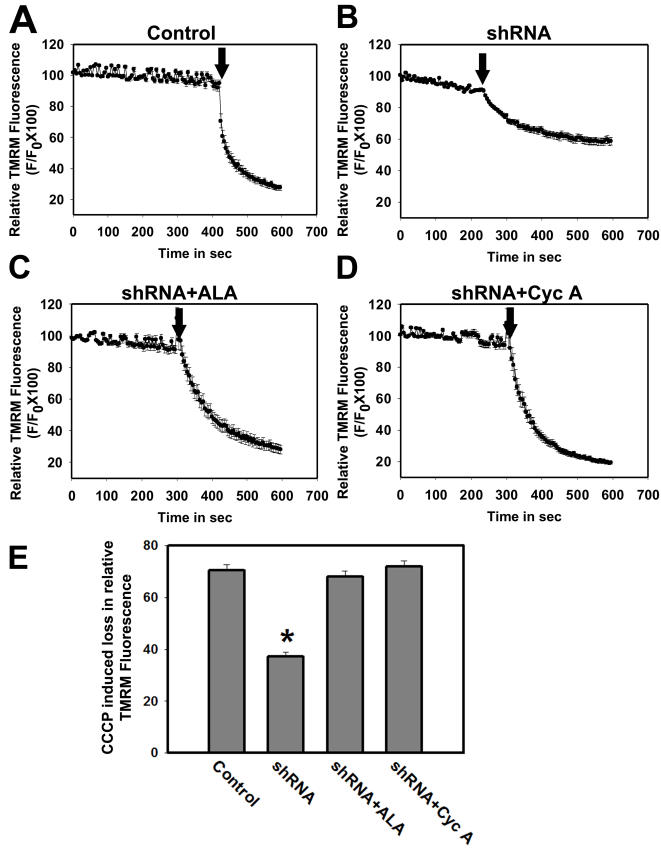
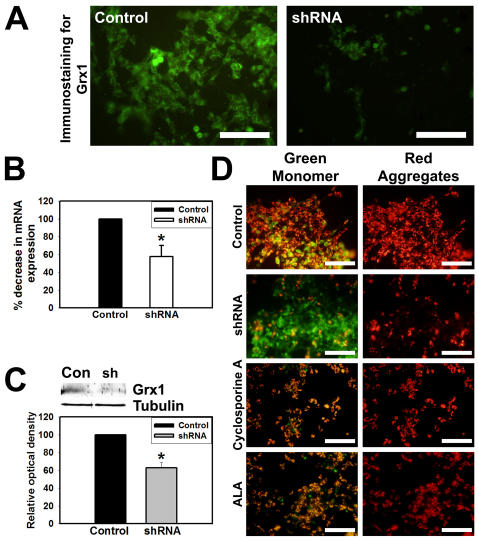
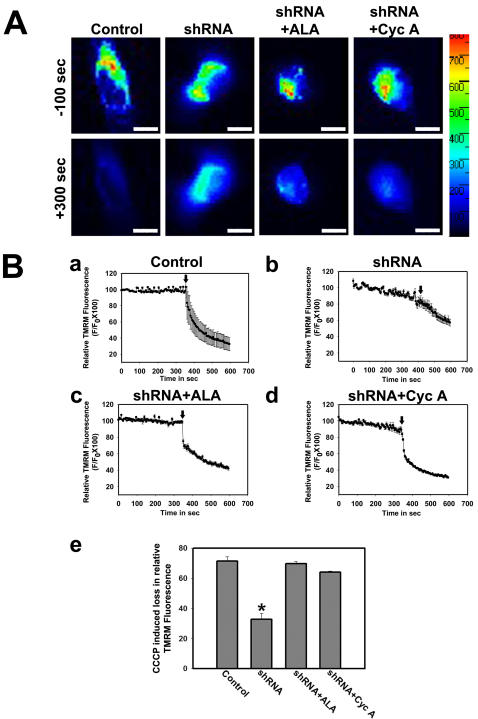
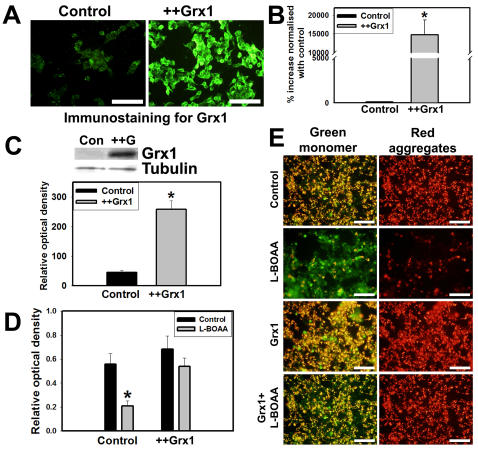
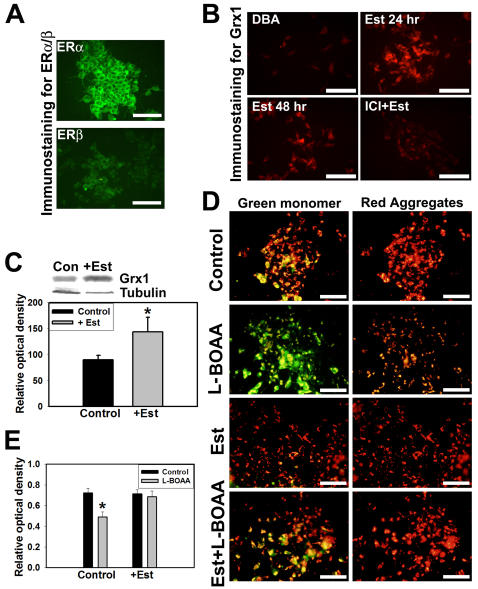
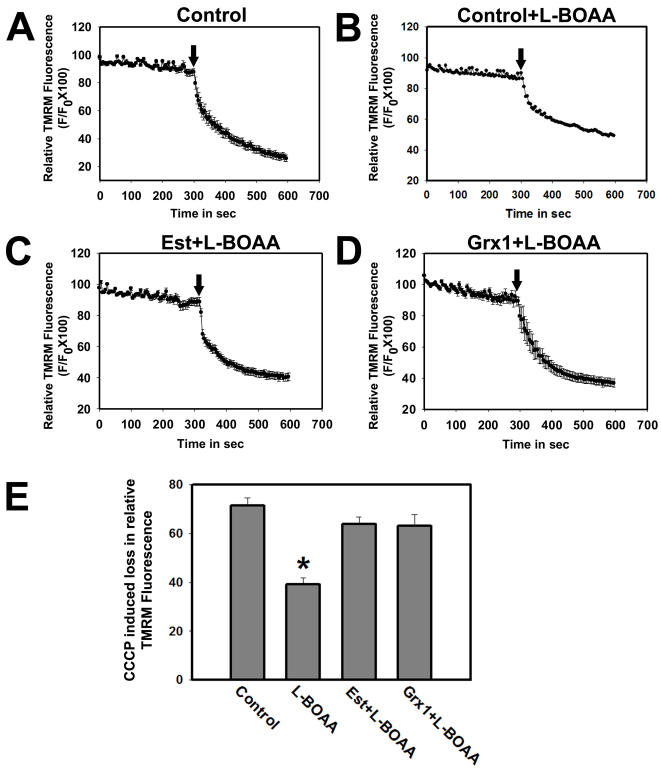
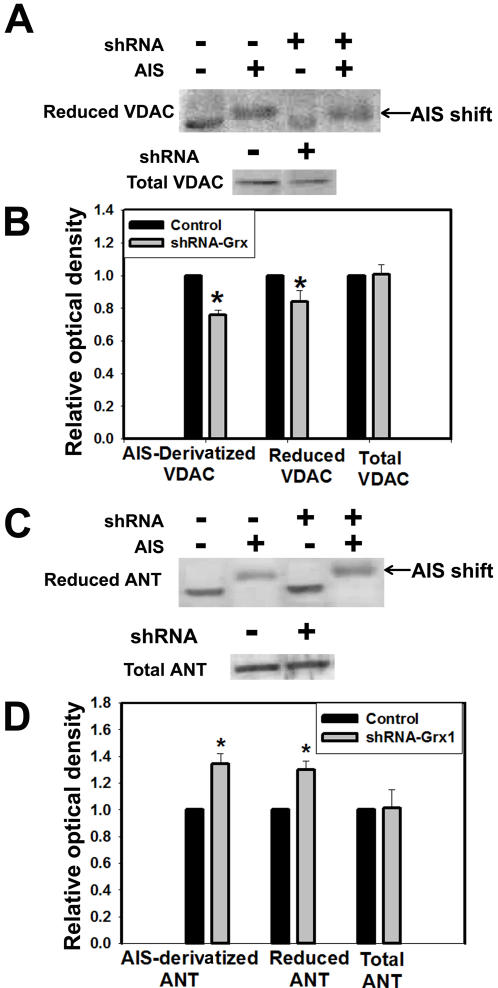
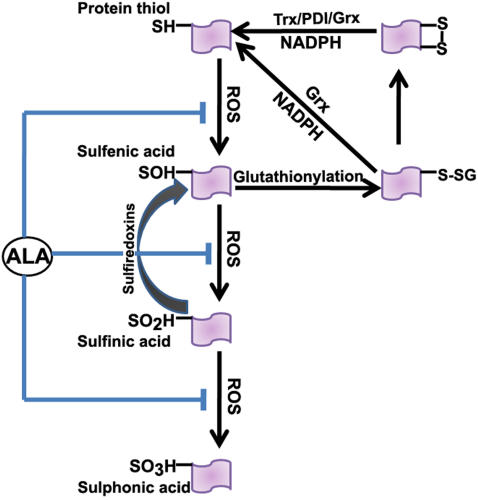
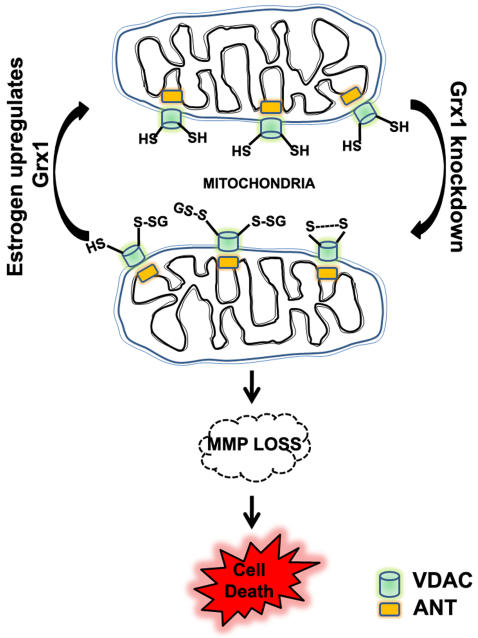
Mitochondrial dysfunction including that caused by oxidative stress has been implicated in the pathogenesis of neurodegenerative diseases. Glutaredoxin 1 (Grx1), a cytosolic thiol disulfide oxido-reductase, reduces glutathionylated proteins to protein thiols and helps maintain redox status of proteins during oxidative stress. Grx1 downregulation aggravates mitochondrial dysfunction in animal models of neurodegenerative diseases, such as Parkinson's and motor neuron disease. We examined the mechanism underlying the regulation of mitochondrial function by Grx1. Downregulation of Grx1 by shRNA results in loss of mitochondrial membrane potential (MMP), which is prevented by the thiol antioxidant, alpha-lipoic acid, or by cyclosporine A, an inhibitor of mitochondrial permeability transition. The thiol groups of voltage dependent anion channel (VDAC), an outer membrane protein in mitochondria but not adenosine nucleotide translocase (ANT), an inner membrane protein, are oxidized when Grx1 is downregulated. We then examined the effect of beta-N-oxalyl amino-L-alanine (L-BOAA), an excitatory amino acid implicated in neurolathyrism (a type of motor neuron disease), that causes mitochondrial dysfunction. Exposure of cells to L-BOAA resulted in loss of MMP, which was prevented by overexpression of Grx1. Grx1 expression is regulated by estrogen in the CNS and treatment of SH-SY5Y cells with estrogen upregulated Grx1 and protected from L-BOAA mediated MMP loss. Our studies demonstrate that Grx1, a cytosolic oxido-reductase, helps maintain mitochondrial integrity and prevents MMP loss caused by oxidative insult. Further, downregulation of Grx1 leads to mitochondrial dysfunction through oxidative modification of the outer membrane protein, VDAC, providing support for the critical role of Grx1 in maintenance of MMP.
Conflict of interest statement
Figures
References
-
- Bambrick LL, Fiskum G. Mitochondrial dysfunction in mouse trisomy 16 brain. Brain Res. 2008;1188:9–16. - PubMed
-
- Orth M, Schapira AH. Mitochondria and degenerative disorders. Am J Med Genet. 2001;106:27–36. - PubMed
-
- Solans A, Zambrano A, Rodriguez M, Barrientos A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum Mol Genet. 2006;15:3063–3081. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
