

Solution structure of the c-terminal dimerization domain of SARS coronavirus nucleocapsid protein solved by the SAIL-NMR method
- PMID: 18561946
- PMCID: PMC7094413
- DOI: 10.1016/j.jmb.2007.11.093
Solution structure of the c-terminal dimerization domain of SARS coronavirus nucleocapsid protein solved by the SAIL-NMR method
Abstract
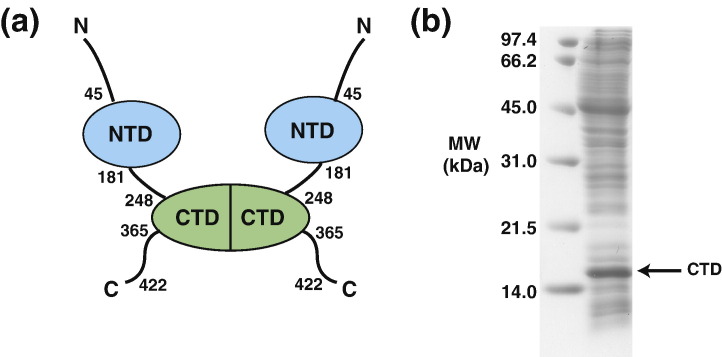
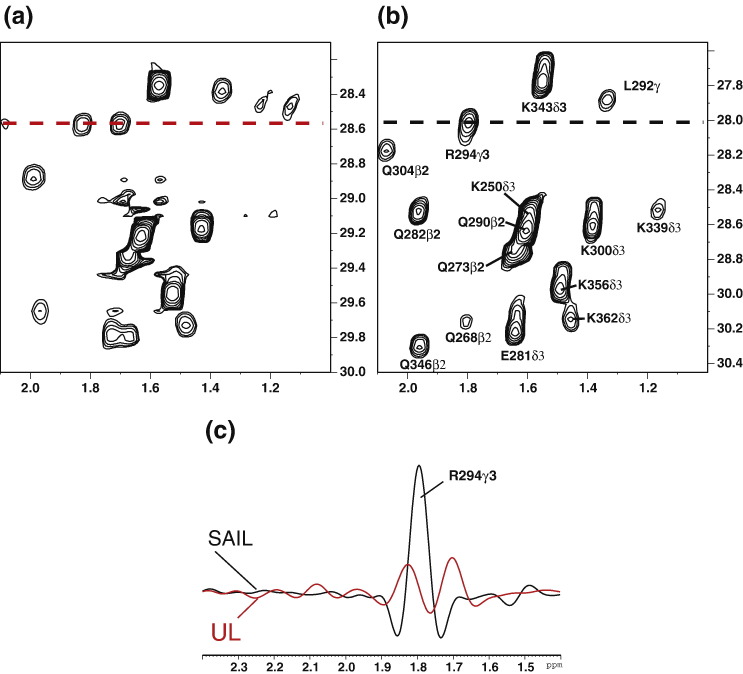
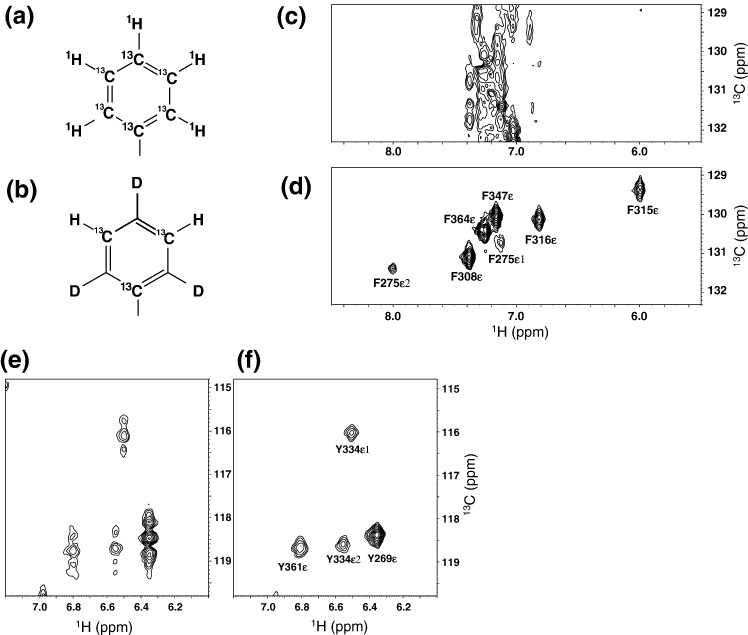
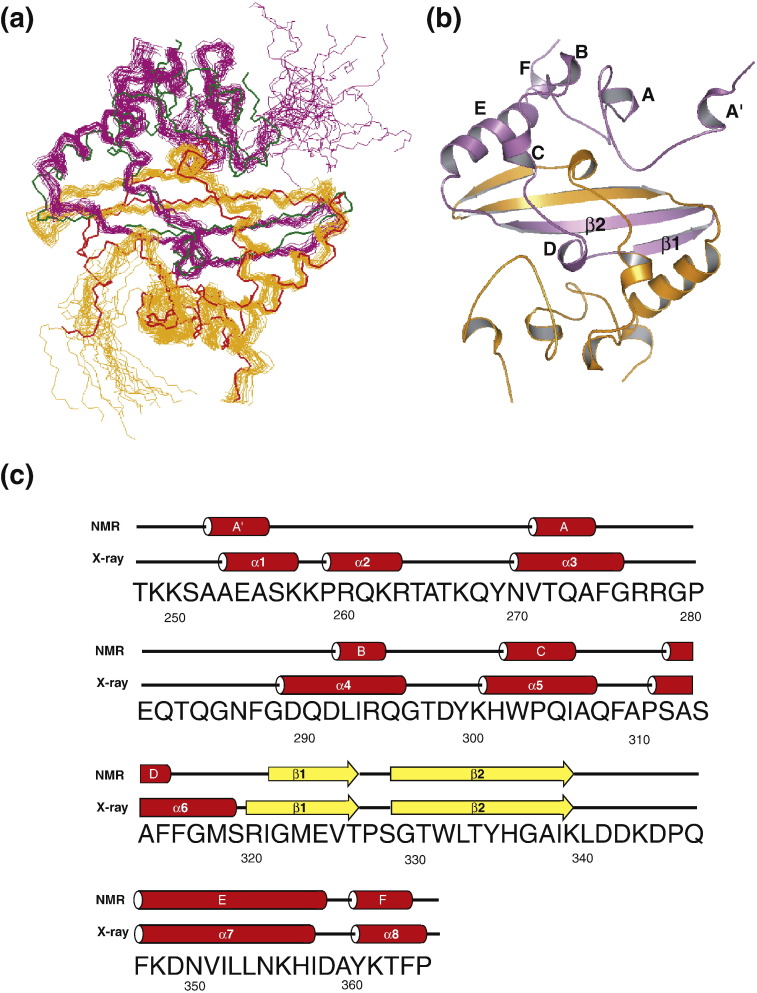
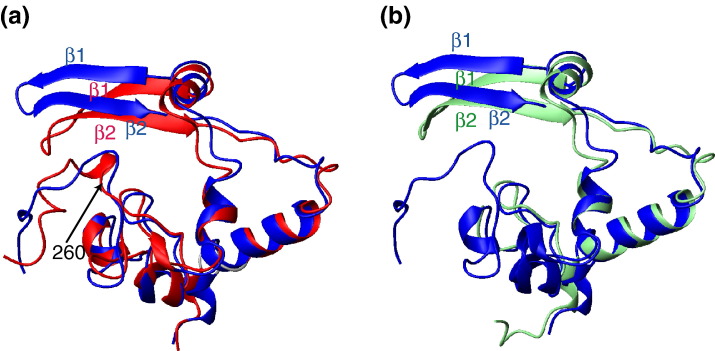
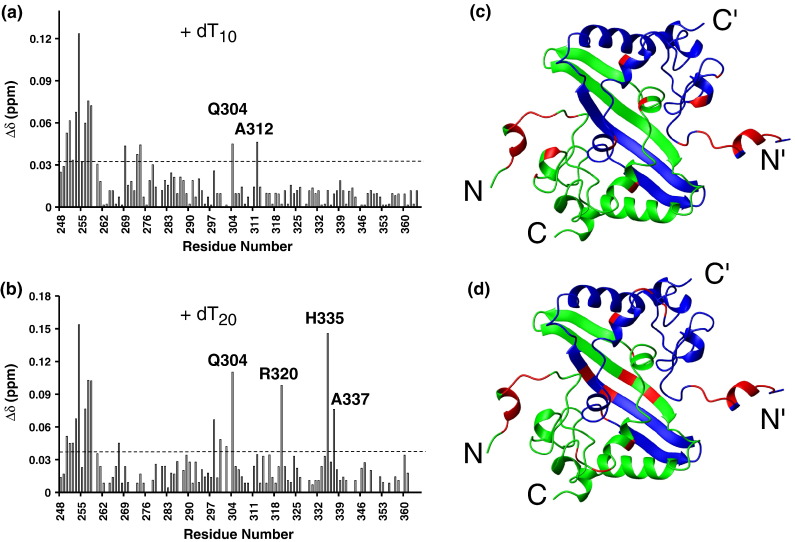
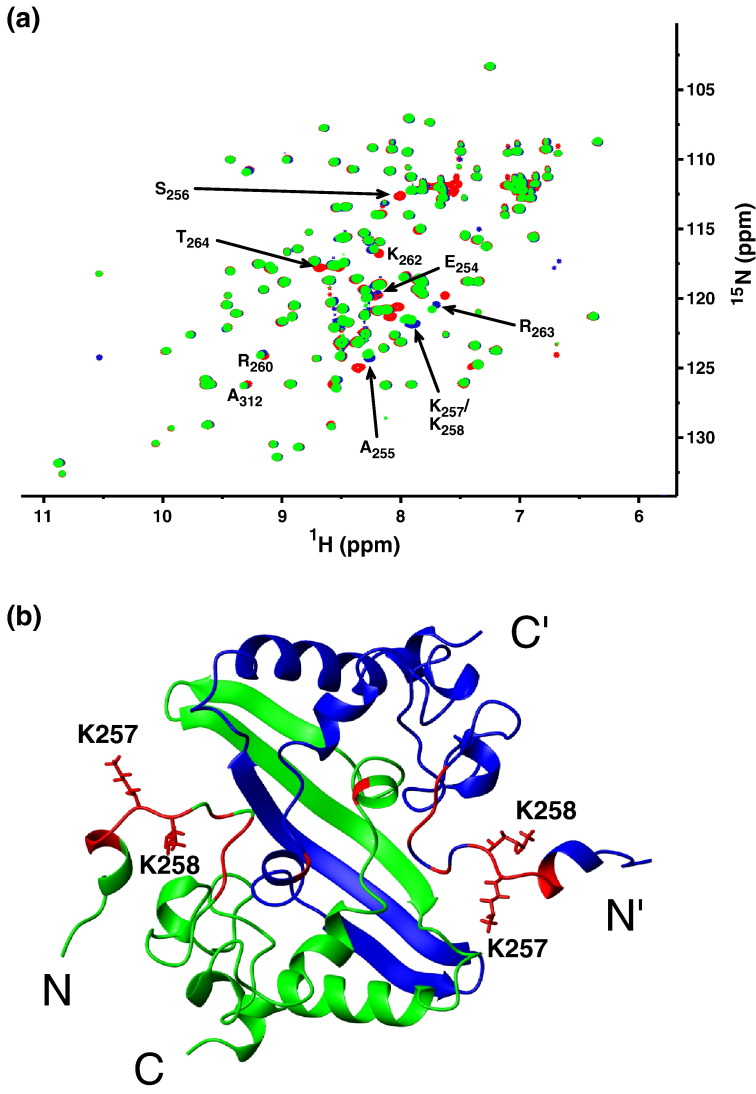
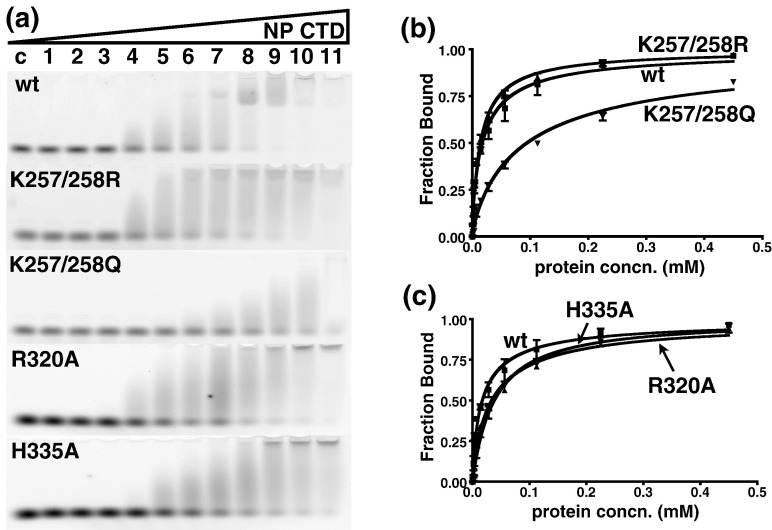
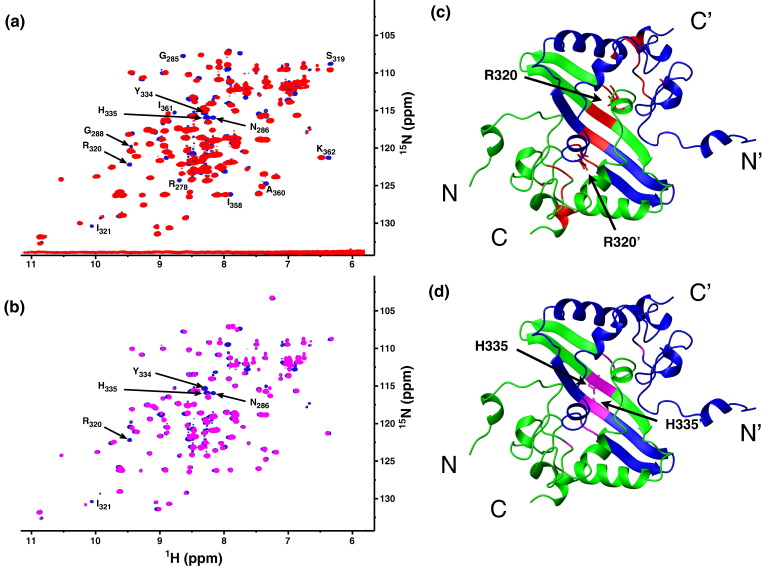
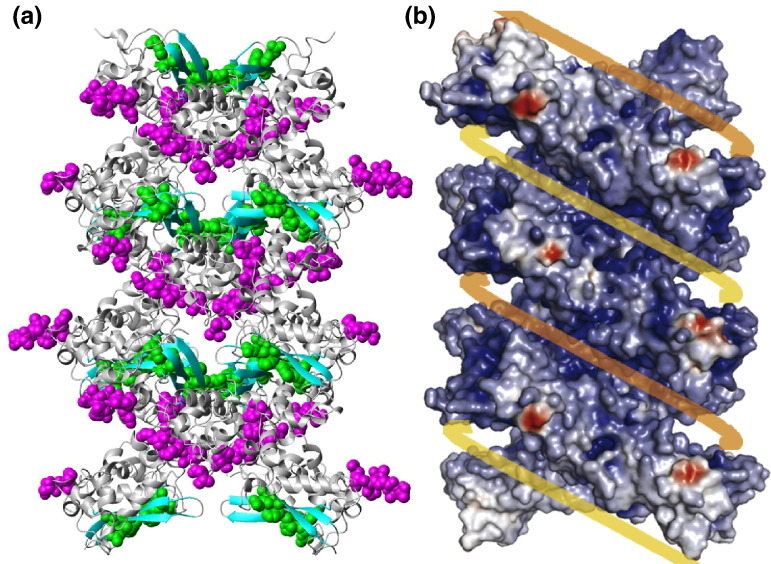
The C-terminal domain (CTD) of the severe acute respiratory syndrome coronavirus (SARS-CoV) nucleocapsid protein (NP) contains a potential RNA-binding region in its N-terminal portion and also serves as a dimerization domain by forming a homodimer with a molecular mass of 28 kDa. So far, the structure determination of the SARS-CoV NP CTD in solution has been impeded by the poor quality of NMR spectra, especially for aromatic resonances. We have recently developed the stereo-array isotope labeling (SAIL) method to overcome the size problem of NMR structure determination by utilizing a protein exclusively composed of stereo- and regio-specifically isotope-labeled amino acids. Here, we employed the SAIL method to determine the high-quality solution structure of the SARS-CoV NP CTD by NMR. The SAIL protein yielded less crowded and better resolved spectra than uniform (13)C and (15)N labeling, and enabled the homodimeric solution structure of this protein to be determined. The NMR structure is almost identical with the previously solved crystal structure, except for a disordered putative RNA-binding domain at the N-terminus. Studies of the chemical shift perturbations caused by the binding of single-stranded DNA and mutational analyses have identified the disordered region at the N-termini as the prime site for nucleic acid binding. In addition, residues in the beta-sheet region also showed significant perturbations. Mapping of the locations of these residues onto the helical model observed in the crystal revealed that these two regions are parts of the interior lining of the positively charged helical groove, supporting the hypothesis that the helical oligomer may form in solution.
Figures
References
-
- Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. - PubMed
-
- Ksiazek T., Erdman D., Goldsmith C., Zaki S., Peret T., Emery S., et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. - PubMed
-
- Huang Q., Yu L., Petros A.M., Gunasekera A., Liu Z., Xu N., et al. Structure of the N-terminal RNA-binding domain of the SARS CoV nucleocapsid protein. Biochemistry. 2004;43:6059–6063. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
