

Conserved footprints of APOBEC3G on Hypermutated human immunodeficiency virus type 1 and human endogenous retrovirus HERV-K(HML2) sequences
- PMID: 18562517
- PMCID: PMC2519685
- DOI: 10.1128/JVI.00584-08
Conserved footprints of APOBEC3G on Hypermutated human immunodeficiency virus type 1 and human endogenous retrovirus HERV-K(HML2) sequences
Abstract
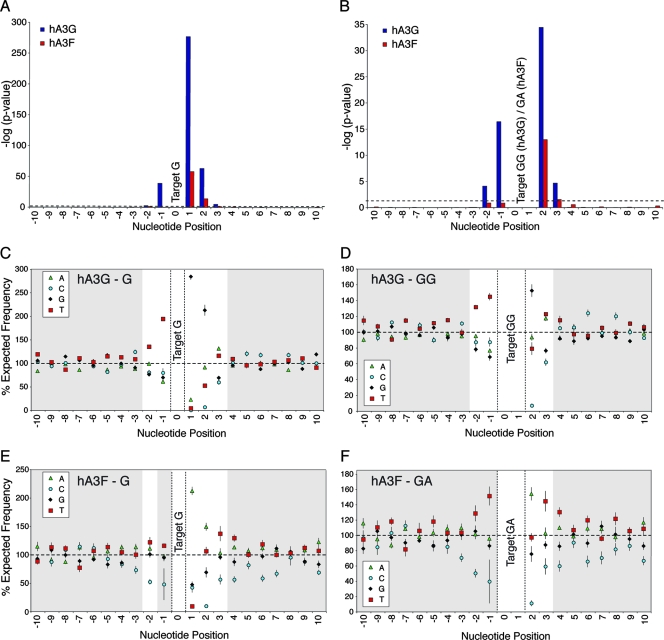
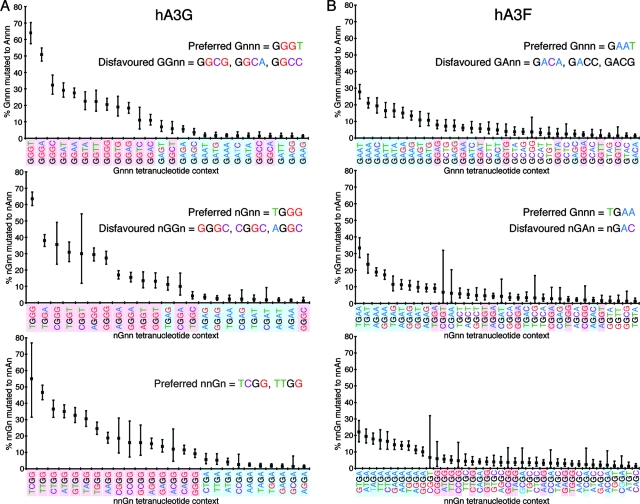
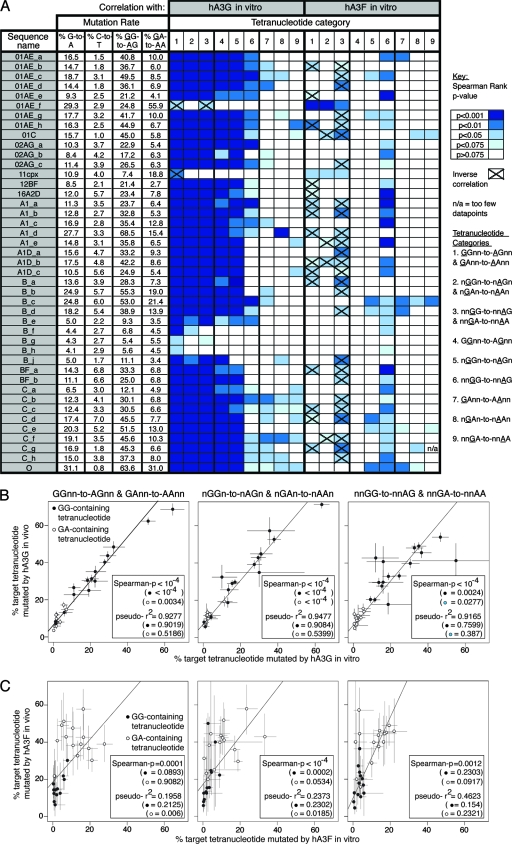
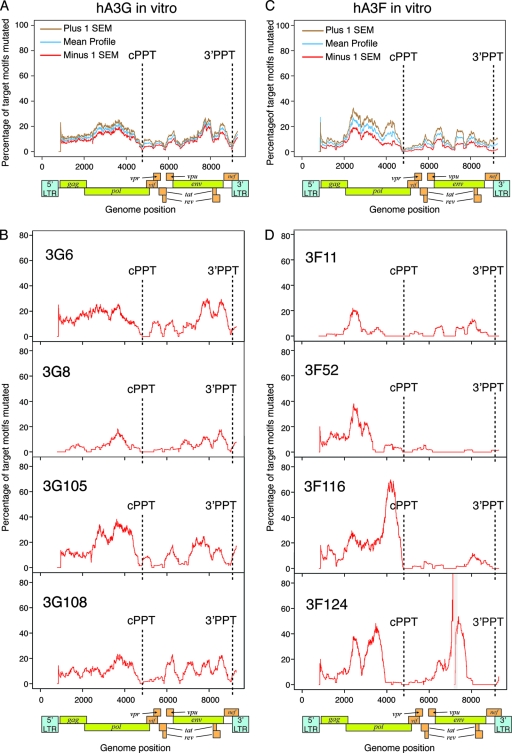
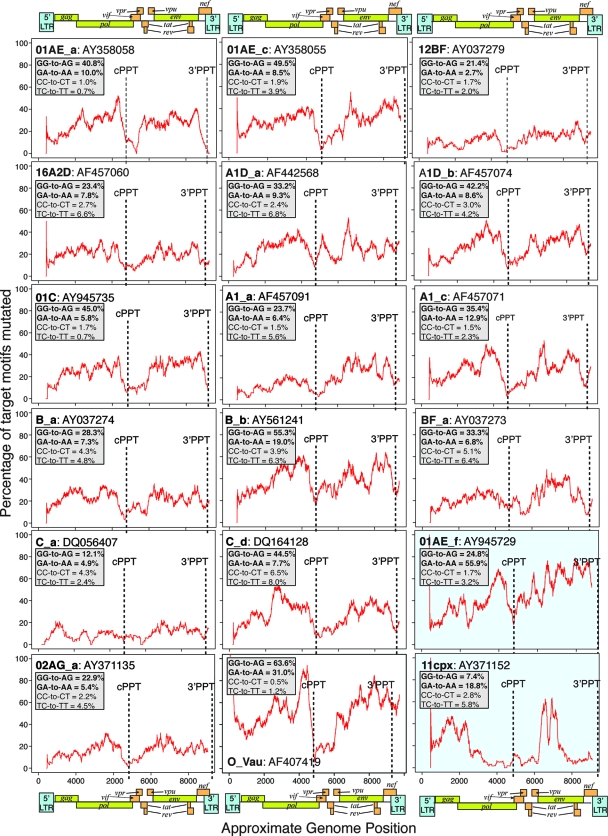
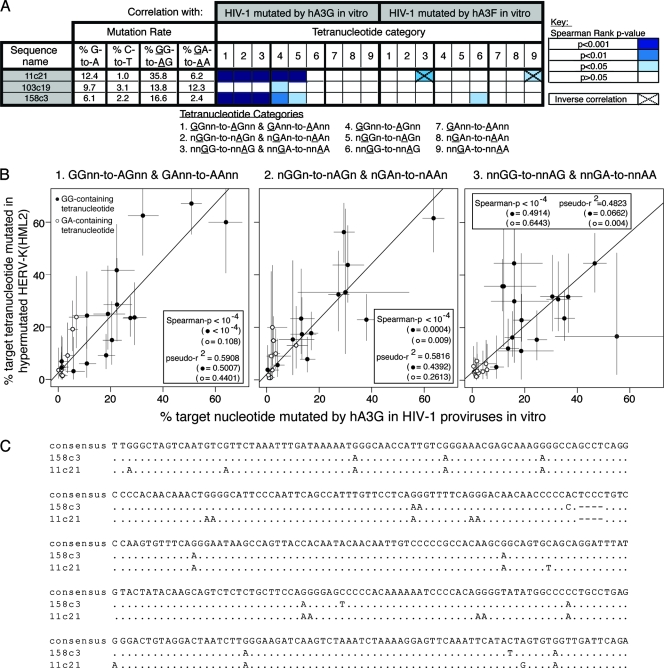
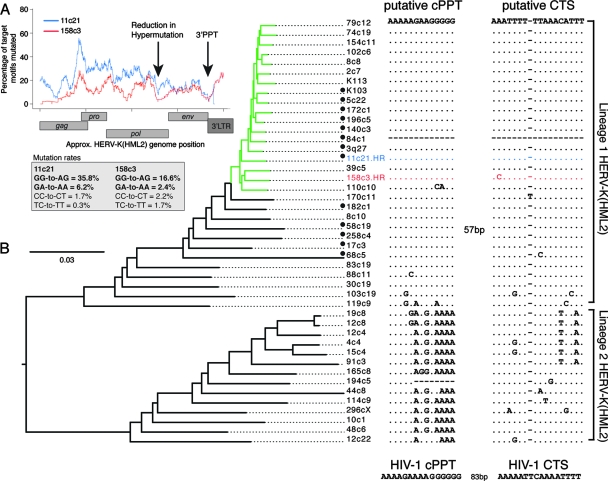
The human polynucleotide cytidine deaminases APOBEC3G (hA3G) and APOBEC3F (hA3F) are antiviral restriction factors capable of inducing extensive plus-strand guanine-to-adenine (G-to-A) hypermutation in a variety of retroviruses and retroelements, including human immunodeficiency virus type 1 (HIV-1). They differ in target specificity, favoring plus-strand 5'GG and 5'GA dinucleotide motifs, respectively. To characterize their mutational preferences in detail, we analyzed single-copy, near-full-length HIV-1 proviruses which had been hypermutated in vitro by hA3G or hA3F. hA3-induced G-to-A mutation rates were significantly influenced by the wider sequence context of the target G. Moreover, hA3G, and to a lesser extent hA3F, displayed clear tetranucleotide preference hierarchies, irrespective of the genomic region examined and overall hypermutation rate. We similarly analyzed patient-derived hypermutated HIV-1 genomes using a new method for estimating reference sequences. The majority of these, regardless of subtype, carried signatures of hypermutation that strongly correlated with those induced in vitro by hA3G. Analysis of genome-wide hA3-induced mutational profiles confirmed that hypermutation levels were reduced downstream of the polypurine tracts. Additionally, while hA3G mutations were found throughout the genome, hA3F often intensely mutated shorter regions, the locations of which varied between proviruses. We extended our analysis to human endogenous retroviruses (HERVs) from the HERV-K(HML2) family, finding two elements that carried clear footprints of hA3G activity. This constitutes the most direct evidence to date for hA3G activity in the context of natural HERV infections, demonstrating the involvement of this restriction factor in defense against retroviral attacks over millions of years of human evolution.
Figures

References
-
- Beale, R. C., S. K. Petersen-Mahrt, I. N. Watt, R. S. Harris, C. Rada, and M. S. Neuberger. 2004. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J. Mol. Biol. 337585-596. - PubMed
-
- Bhattacharya, T., M. Daniels, D. Heckerman, B. Foley, N. Frahm, C. Kadie, J. Carlson, K. Yusim, B. McMahon, B. Gaschen, S. Mallal, J. I. Mullins, D. C. Nickle, J. Herbeck, C. Rousseau, G. H. Learn, T. Miura, C. Brander, B. Walker, and B. Korber. 2007. Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science 3151583-1586. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
