

Testing for neutrality in samples with sequencing errors
- PMID: 18562660
- PMCID: PMC2475743
- DOI: 10.1534/genetics.107.082198
Testing for neutrality in samples with sequencing errors
Abstract
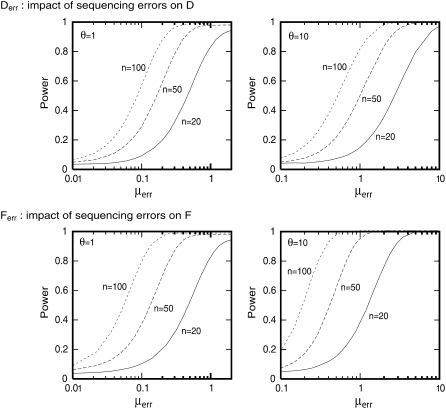
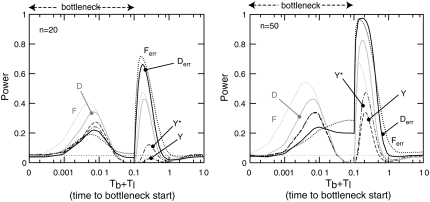
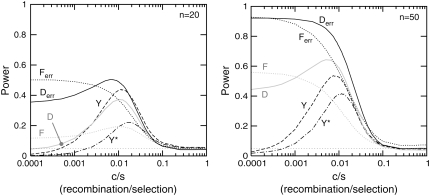
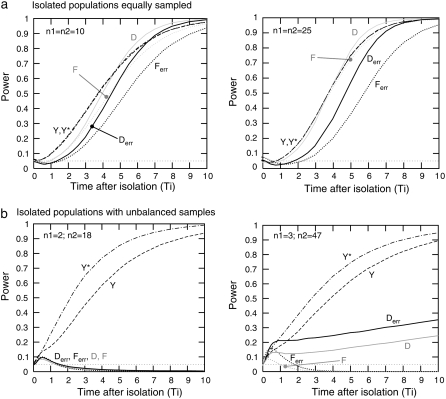
Many data sets one could use for population genetics contain artifactual sites, i.e., sequencing errors. Here, we first explore the impact of such errors on several common summary statistics, assuming that sequencing errors are mostly singletons. We thus show that in the presence of those errors, estimators of can be strongly biased. We further show that even with a moderate number of sequencing errors, neutrality tests based on the frequency spectrum reject neutrality. This implies that analyses of data sets with such errors will systematically lead to wrong inferences of evolutionary scenarios. To avoid to these errors, we propose two new estimators of theta that ignore singletons as well as two new tests Y and Y* that can be used to test neutrality despite sequencing errors. All in all, we show that even though singletons are ignored, these new tests show some power to detect deviations from a standard neutral model. We therefore advise the use of these new tests to strengthen conclusions in suspicious data sets.
Figures
References
-
- Achaz, G., S. Palmer, M. Kearney, F. Maldarelli, J. W. Mellors et al., 2004. A robust measure of HIV-1 population turnover within chronically infected individuals. Mol. Biol. Evol. 21 1902–1912. - PubMed
-
- Berger, R., and D. Boos, 1994. P values maximized over a confidence set for the nuisance parameter. J. Am. Stat. Assoc. 89 1012–1016.
-
- Depaulis, F., and M. Veuille, 1998. Neutrality tests based on the distribution of haplotypes under an infinite-site model. Mol. Biol. Evol. 15 1788–1790. - PubMed
-
- Depaulis, F., S. Mousset and M. Veuille, 2001. Haplotype tests using coalescent simulations conditional on the number of segregating sites. Mol. Biol. Evol. 18 1136–1138. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
