

Discovery and verification of functional single nucleotide polymorphisms in regulatory genomic regions: current and developing technologies
- PMID: 18565787
- PMCID: PMC2676583
- DOI: 10.1016/j.mrrev.2008.05.001
Discovery and verification of functional single nucleotide polymorphisms in regulatory genomic regions: current and developing technologies
Abstract
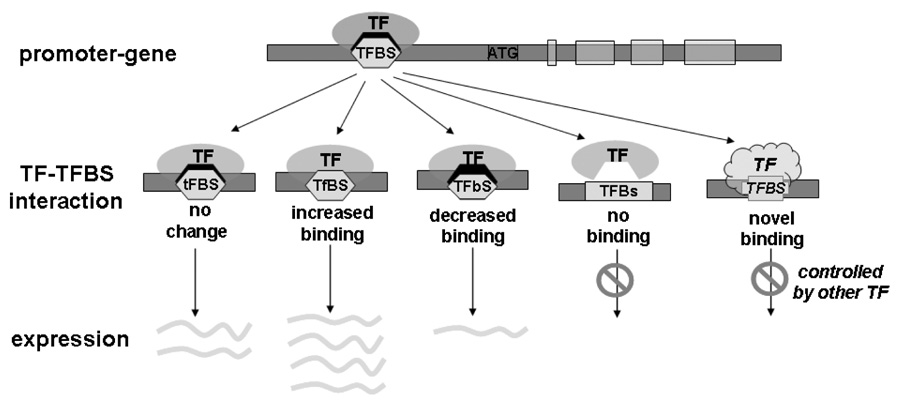
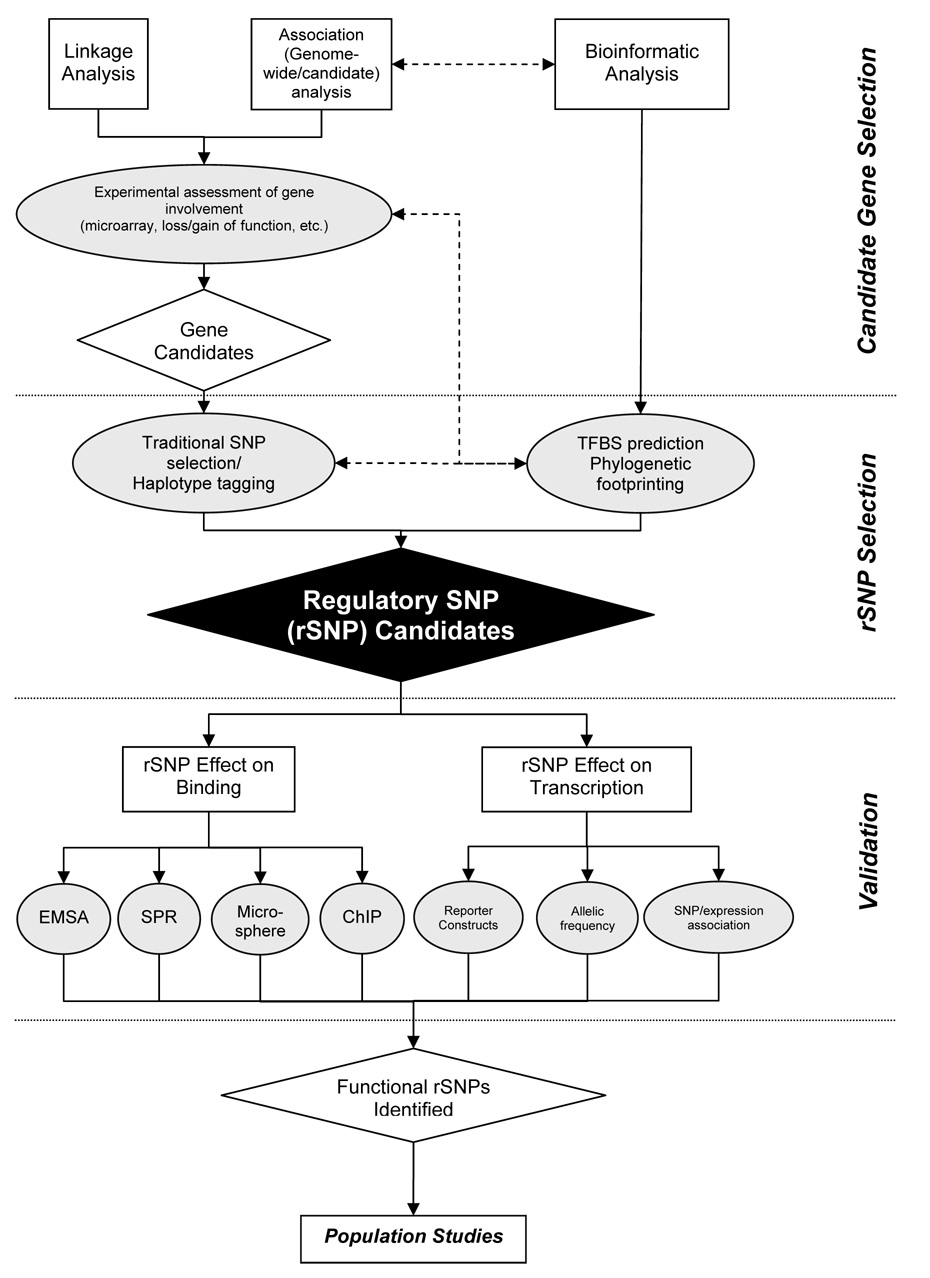
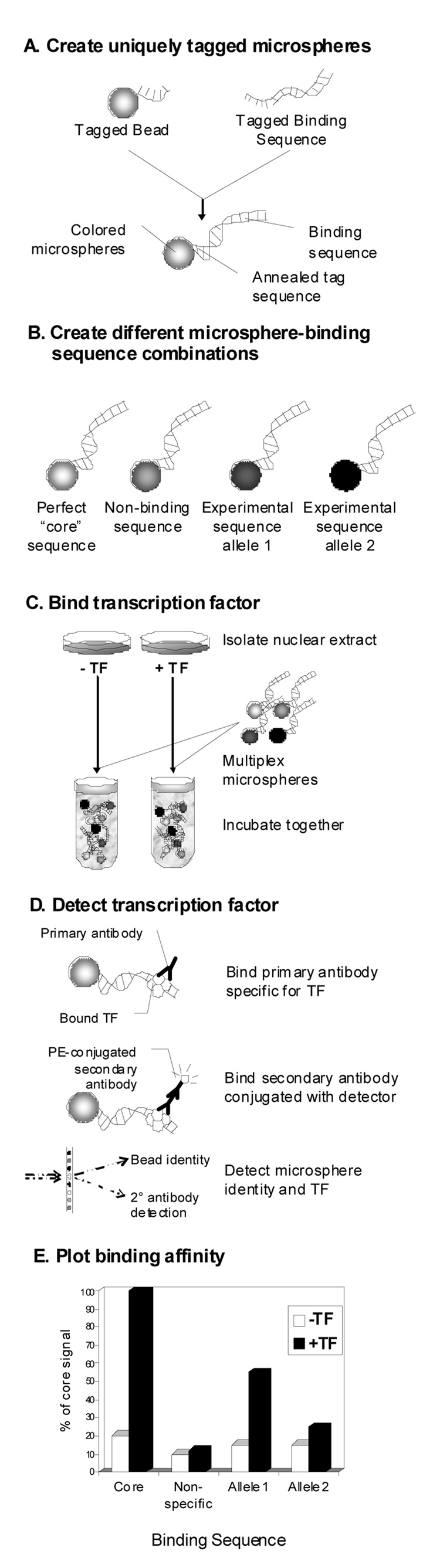
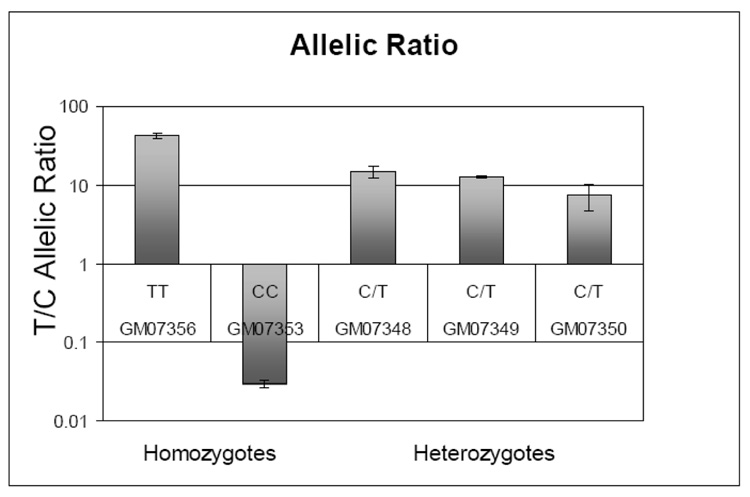
The most common form of genetic variation, single nucleotide polymorphisms or SNPs, can affect the way an individual responds to the environment and modify disease risk. Although most of the millions of SNPs have little or no effect on gene regulation and protein activity, there are many circumstances where base changes can have deleterious effects. Non-synonymous SNPs that result in amino acid changes in proteins have been studied because of their obvious impact on protein activity. It is well known that SNPs within regulatory regions of the genome can result in disregulation of gene transcription. However, the impact of SNPs located in putative regulatory regions, or rSNPs, is harder to predict for two primary reasons. First, the mechanistic roles of non-coding genomic sequence remain poorly defined. Second, experimental validation of the functional consequences of rSNPs is often slow and laborious. In this review, we summarize traditional and novel methodologies for candidate rSNPs selection, in particular in silico techniques that aid in candidate rSNP selection. Additionally we will discuss molecular biological techniques that assess the impact of rSNPs on binding of regulatory machinery, as well as functional consequences on transcription. Standard techniques such as EMSA and luciferase reporter constructs are still widely used to assess effects of rSNPs on binding and gene transcription; however, these protocols are often bottlenecks in the discovery process. Therefore, we highlight novel and developing high-throughput protocols that promise to aid in shortening the process of rSNP validation. Given the large amount of genomic information generated from a multitude of re-sequencing and genome-wide SNP array efforts, future focus should be to develop validation techniques that will allow greater understanding of the impact these polymorphisms have on human health and disease.
Figures
References
-
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey DL, Hunt SE, Cole CG, Coggill PC, Rice CM, Ning Z, Rogers J, Bentley DR, Kwok PY, Mardis ER, Yeh RT, Schultz B, Cook L, Davenport R, Dante M, Fulton L, Hillier L, Waterston RH, McPherson JD, Gilman B, Schaffner S, Van Etten WJ, Reich D, Higgins J, Daly MJ, Blumenstiel B, Baldwin J, Stange-Thomann N, Zody MC, Linton L, Lander ES, Altshuler D. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. - PubMed
-
- Collins FS, Brooks LD, Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998;8:1229–1231. - PubMed
-
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
-
- I.H.G.S. Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. - PubMed
-
- Ponomarenko JV, Orlova GV, Merkulova TI, Gorshkova EV, Fokin ON, Vasiliev GV, Frolov AS, Ponomarenko MP. rSNP_Guide: an integrated database-tools system for studying SNPs and site-directed mutations in transcription factor binding sites. Hum Mutat. 2002;20:239–248. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
