

Phenylketonuria: an inborn error of phenylalanine metabolism
- PMID: 18566668
- PMCID: PMC2423317
Phenylketonuria: an inborn error of phenylalanine metabolism
Abstract
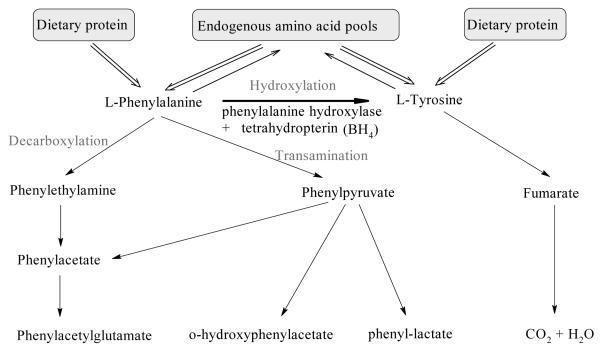
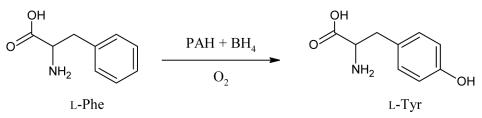
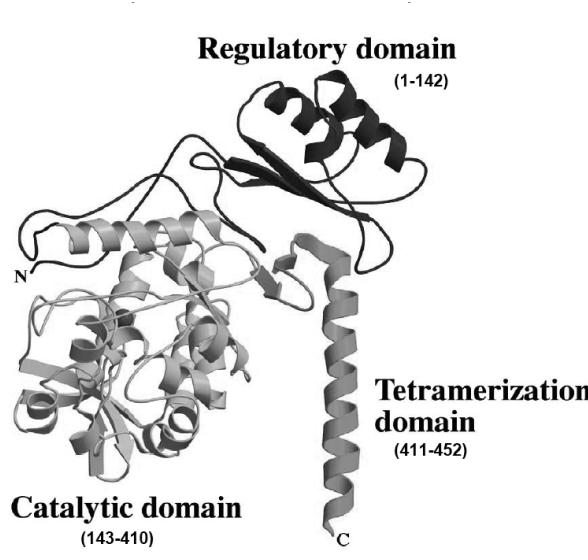
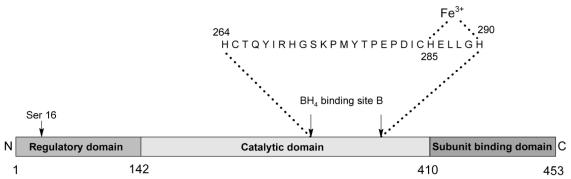
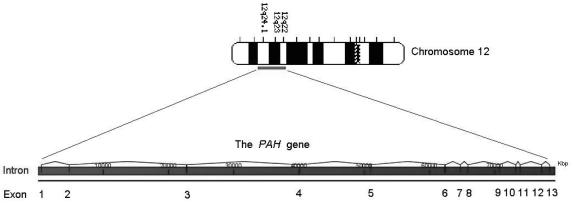
Phenylketonuria (PKU) is an autosomal recessive inborn error of phenylalanine (Phe) metabolism resulting from deficiency of phenylalanine hydroxylase (PAH). Most forms of PKU and hyperphenylalaninaemia (HPA) are caused by mutations in the PAH gene on chromosome 12q23.2. Untreated PKU is associated with an abnormal phenotype which includes growth failure, poor skin pigmentation, microcephaly, seizures, global developmental delay and severe intellectual impairment. However, since the introduction of newborn screening programs and with early dietary intervention, children born with PKU can now expect to lead relatively normal lives. A better understanding of the biochemistry, genetics and molecular basis of PKU, as well as the need for improved treatment options, has led to the development of new therapeutic strategies.
Figures
References
-
- Hanley WB. Adult Phenylketonuria. Am J Med. 2004;117:590–5. - PubMed
-
- Centerwall SA, Centerwall WR. The discovery of phenylketonuria: the story of a young couple, two retarded children, and a scientist. Pediatrics. 2000;105:89–103. - PubMed
-
- Følling I. The discovery of phenylketonuria. Acta Pediatr Suppl. 1994;407:4–10. - PubMed
-
- Følling A. Über ausscheidung von phenylbrenztraubensäure in den harn als stoffwechselanomalie in verbindung mit imbezillität. Hoppe-Seylers Z Physiol Chem. 1934;227:169–76.
LinkOut - more resources
Full Text Sources
Medical