

Mitochondrial medicine for aging and neurodegenerative diseases
- PMID: 18566920
- PMCID: PMC3235551
- DOI: 10.1007/s12017-008-8044-z
Mitochondrial medicine for aging and neurodegenerative diseases
Abstract
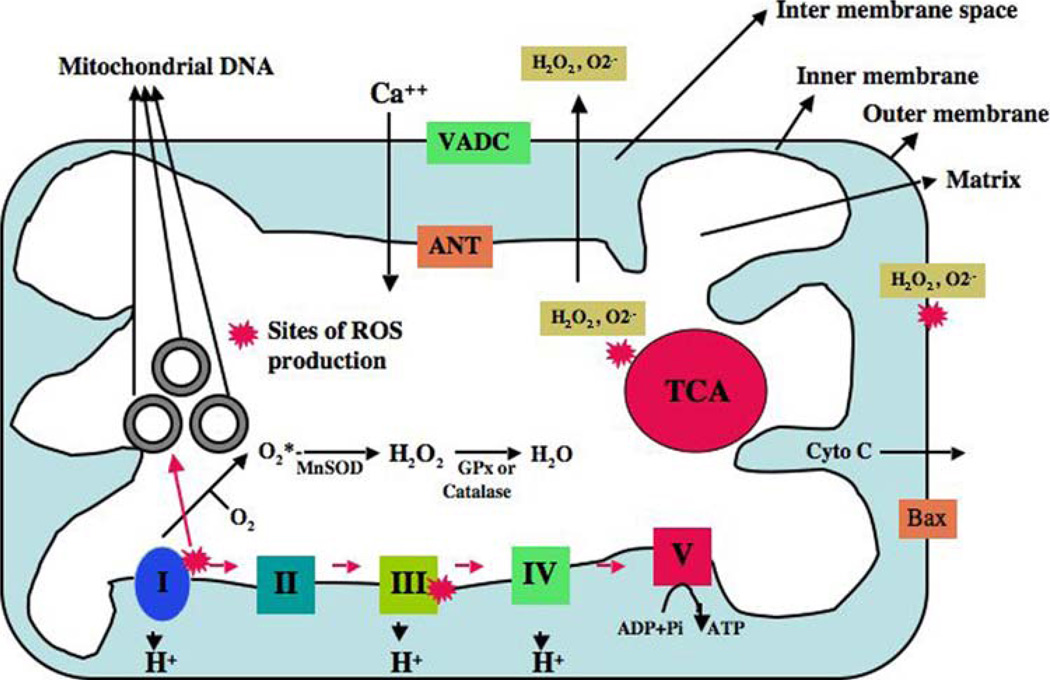
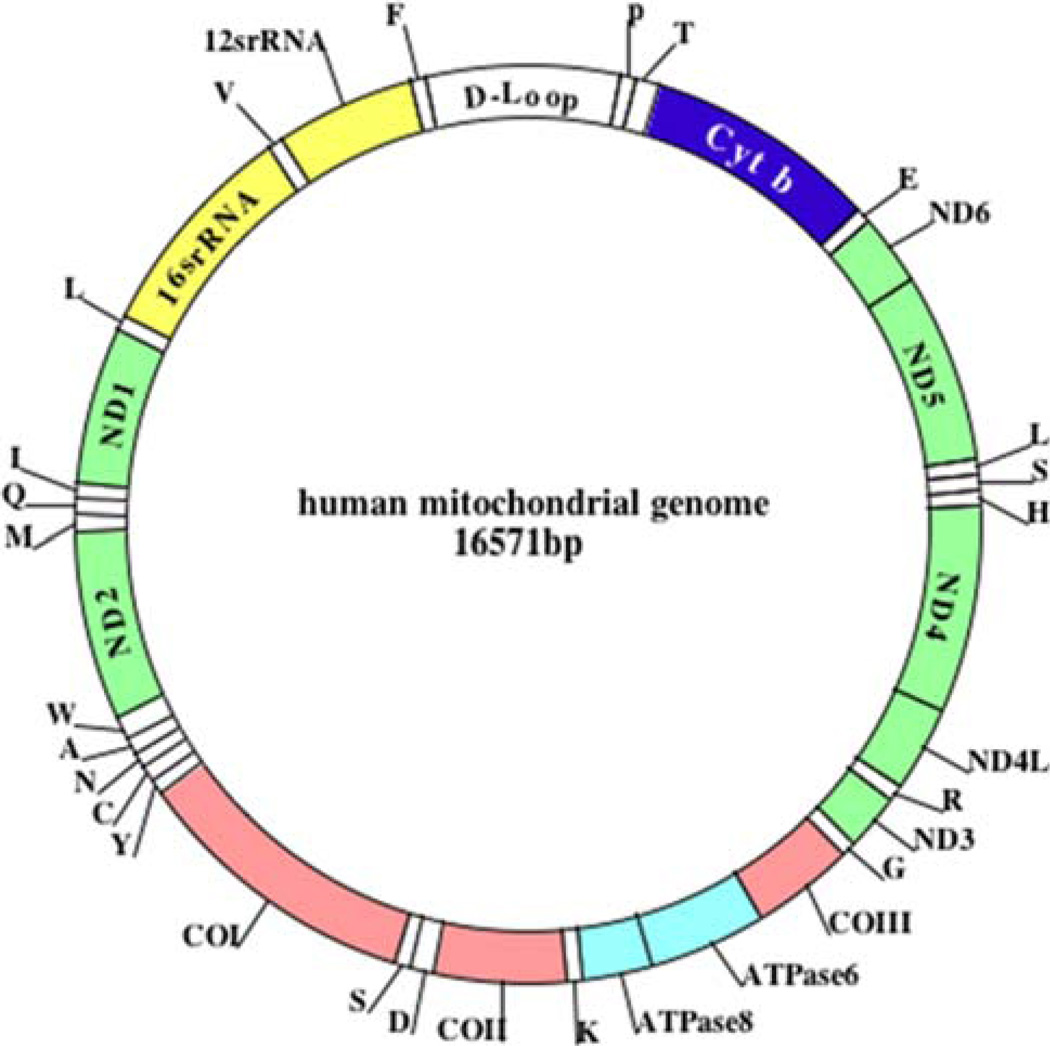
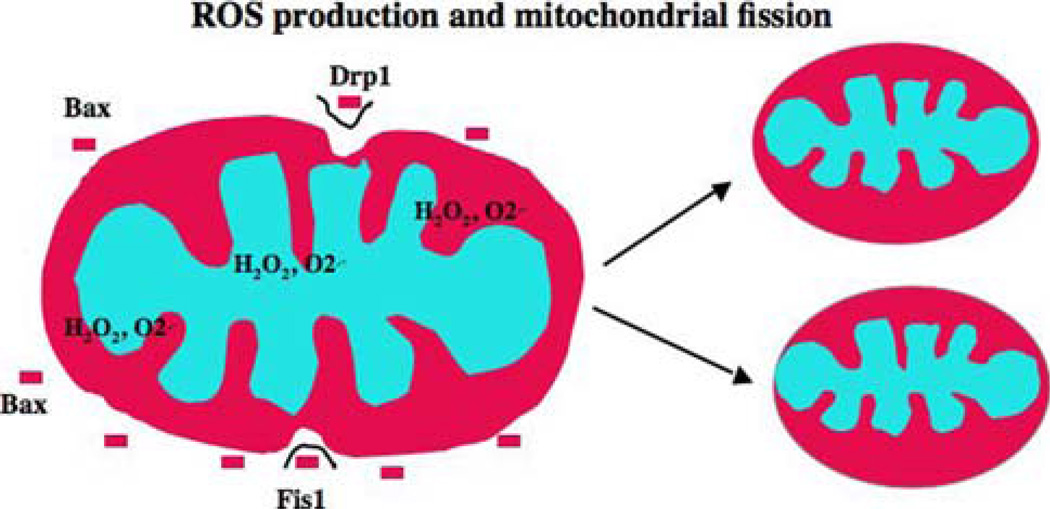
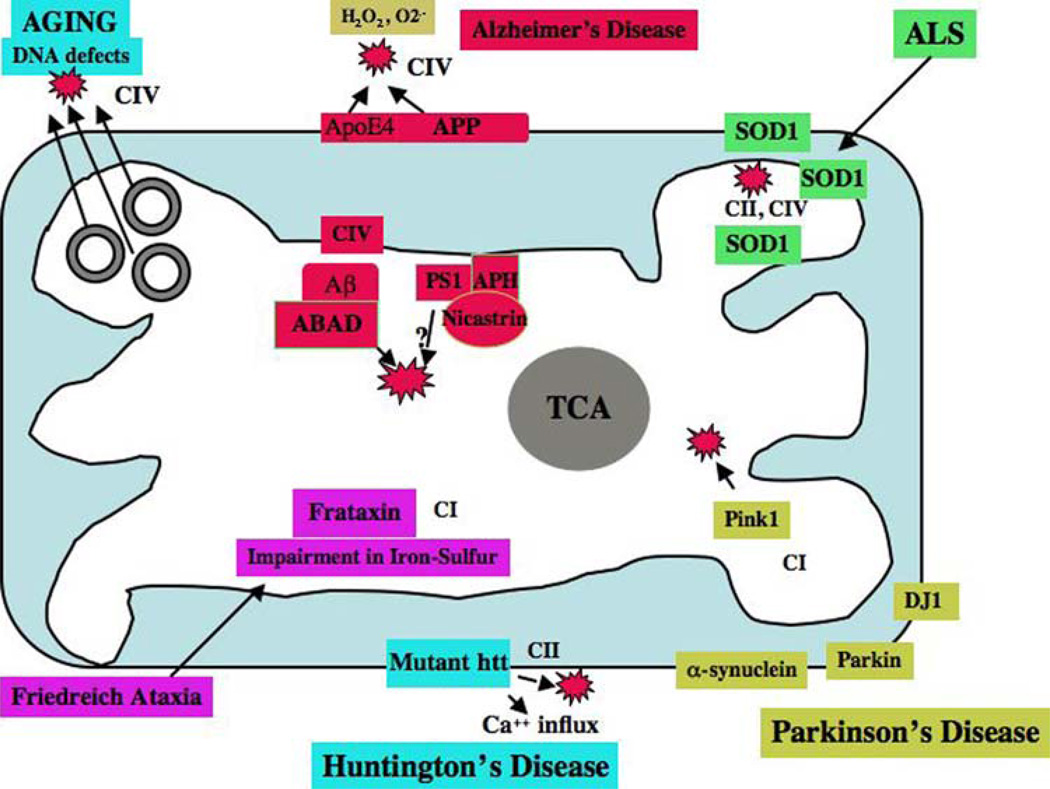
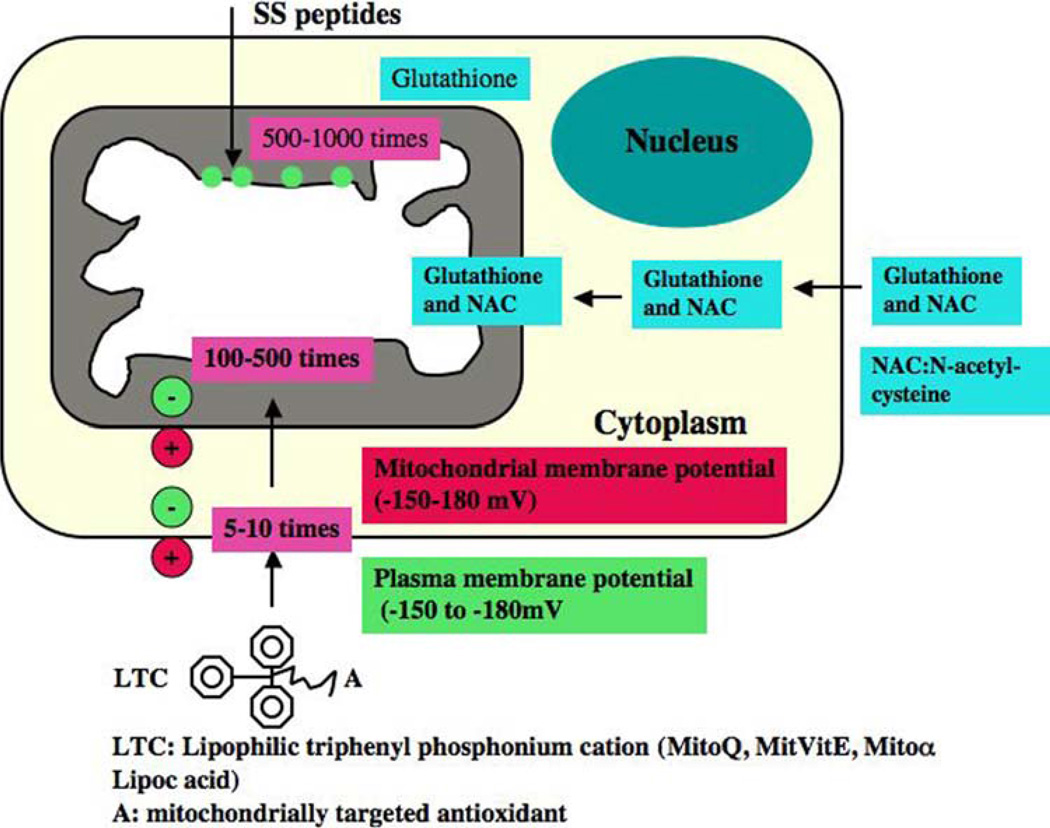
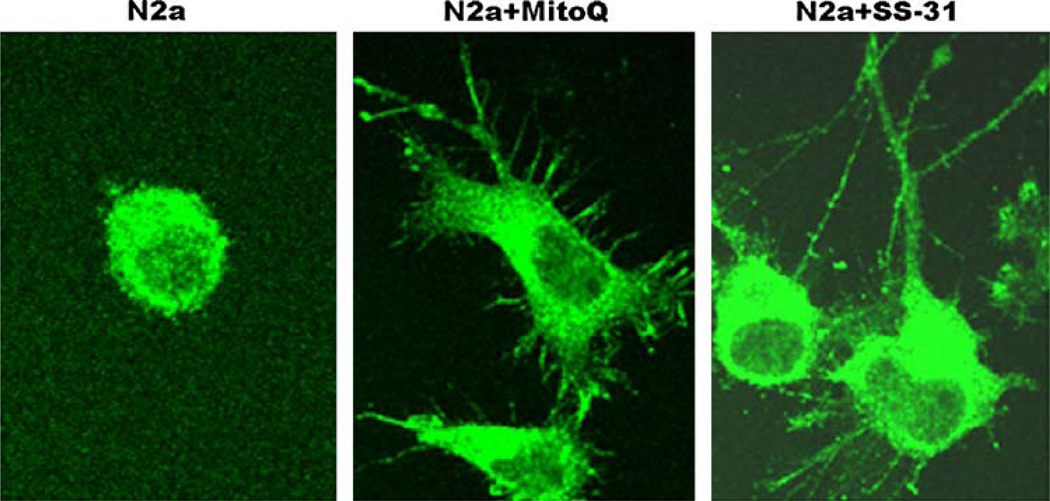
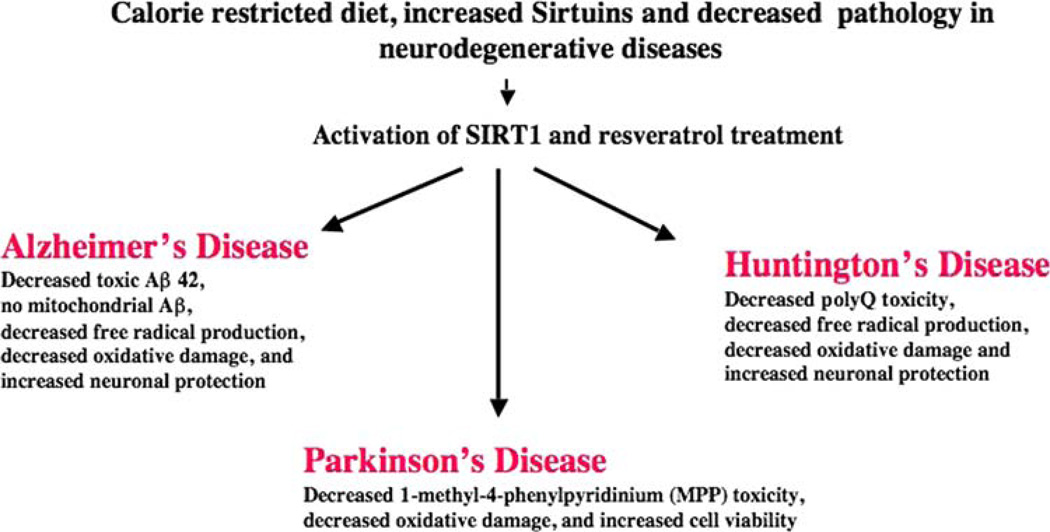
Mitochondria are key cytoplasmic organelles, responsible for generating cellular energy, regulating intracellular calcium levels, altering the reduction-oxidation potential of cells, and regulating cell death. Increasing evidence suggests that mitochondria play a central role in aging and in neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, Huntington's disease, amyotrophic lateral sclerosis, and Freidriech ataxia. Further, several lines of evidence suggest that mitochondrial dysfunction is an early event in most late-onset neurodegenerative diseases. Biochemical and animal model studies of inherited neurodegenerative diseases have revealed that mutant proteins of these diseases are associated with mitochondria. Mutant proteins are reported to block the transport of nuclear-encoded mitochondrial proteins to mitochondria, interact with mitochondrial proteins and disrupt the electron transport chain, induce free radicals, cause mitochondrial dysfunction, and, ultimately, damage neurons. This article discusses critical issues of mitochondria causing dysfunction in aging and neurodegenerative diseases, and discusses the potential of developing mitochondrial medicine, particularly mitochondrially targeted antioxidants, to treat aging and neurodegenerative diseases.
Figures
References
-
- Abe Y, Hashimoto Y, Tomita Y, Terashita K, Aiso S, Tajima H, et al. Cytotoxic mechanisms by M239 V presenilin 2, a little-analyzed Alzheimer’s disease-causative mutant. Journal of Neuroscience Research. 2004;77:583–595. - PubMed
-
- Abeliovich A, Beal MF. Parkinsonism genes: Culprits and clues. Journal of Neurochemistry. 2006;99:1062–1072. - PubMed
-
- Afifi AK, Aleu FP, Goodgold J, MacKay B. Ultrastructure of atrophic muscle in amyotrophic lateral sclerosis. Neurology. 1966;16:475–481. - PubMed
-
- Andersen JK. Iron dysregulation and Parkinson’s disease. Journal of Alzheimer’s Disease. 2004;6:S47–S52. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
