

Multiple signaling pathways are responsible for prostaglandin E2-induced murine keratinocyte proliferation
- PMID: 18567804
- PMCID: PMC2759608
- DOI: 10.1158/1541-7786.MCR-07-2144
Multiple signaling pathways are responsible for prostaglandin E2-induced murine keratinocyte proliferation
Abstract
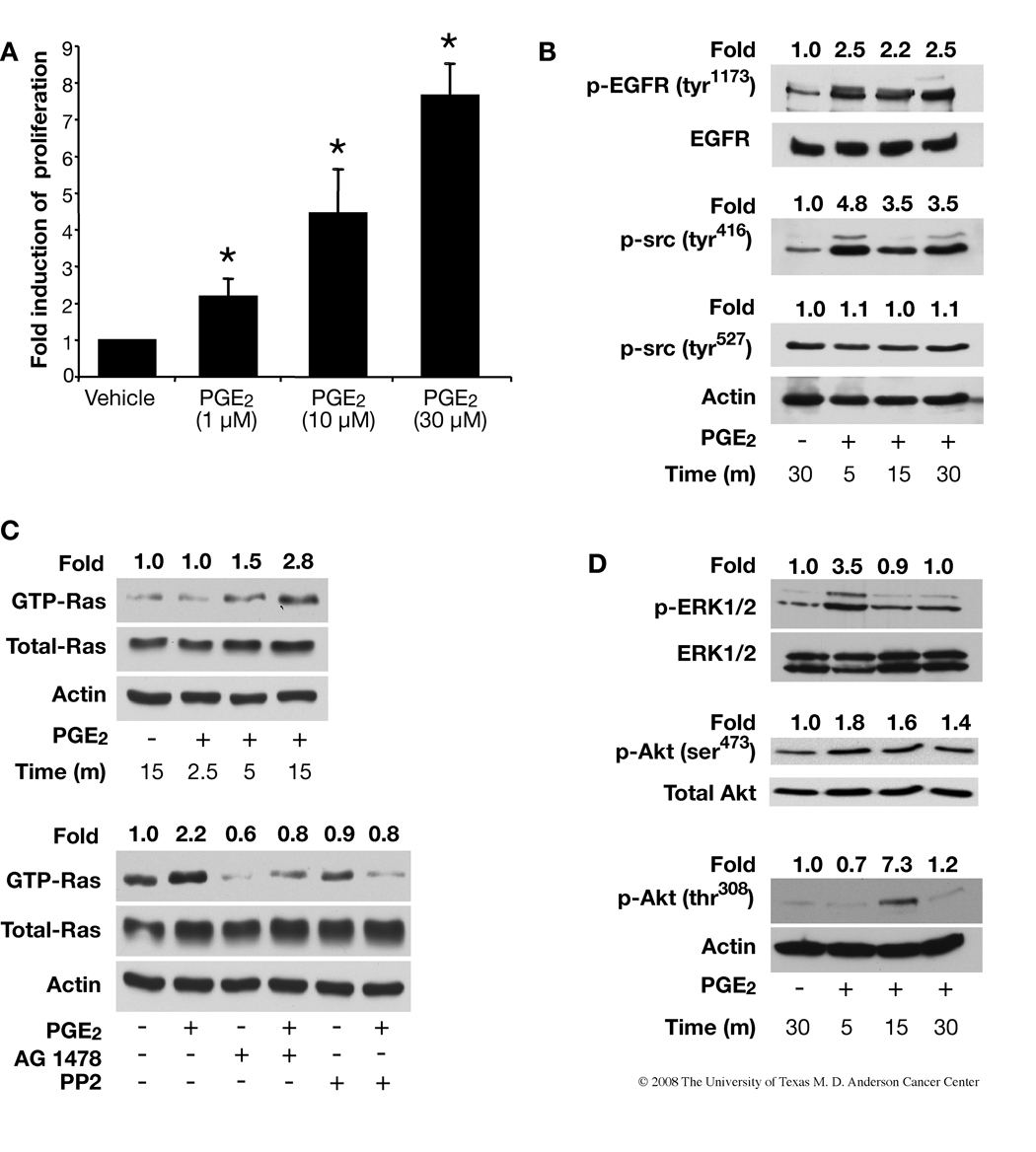
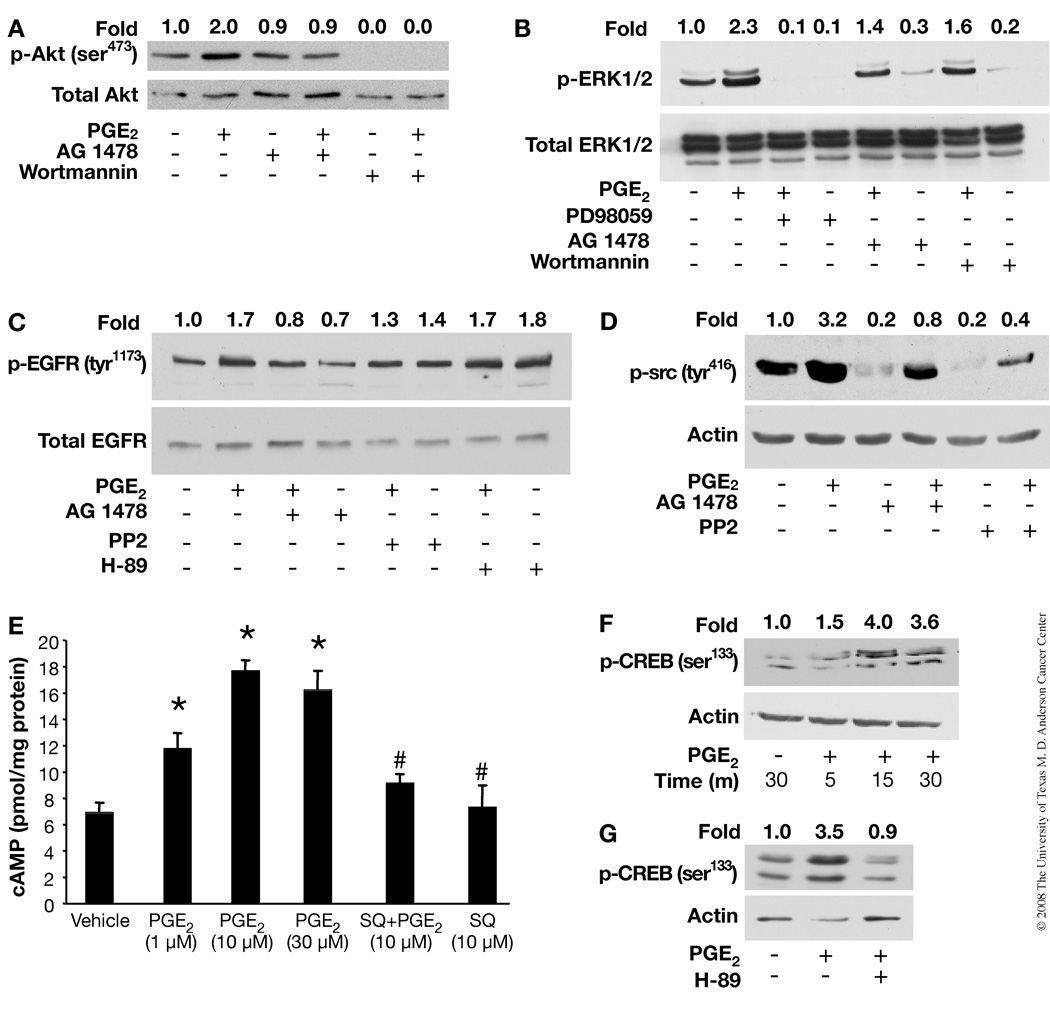
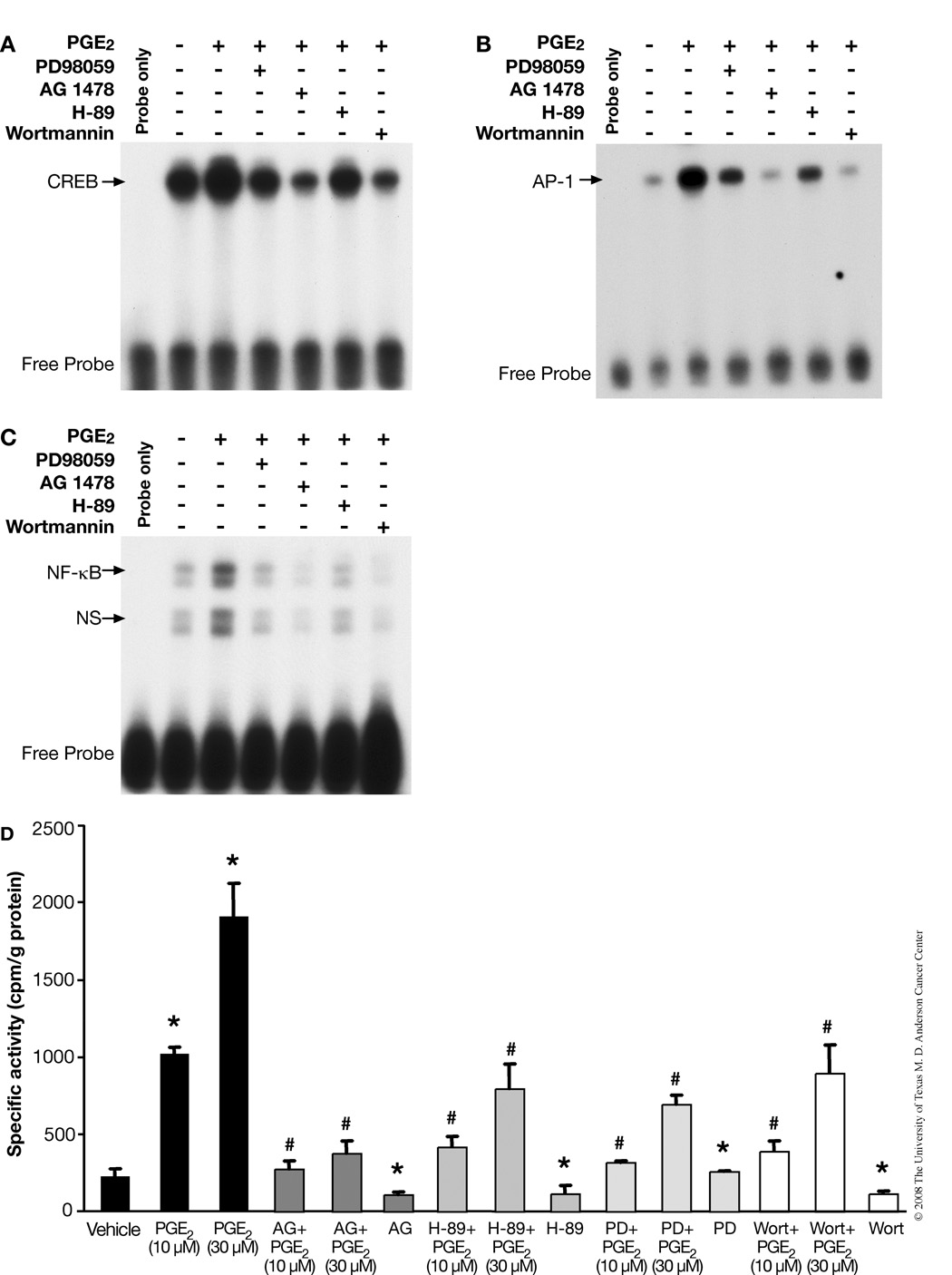
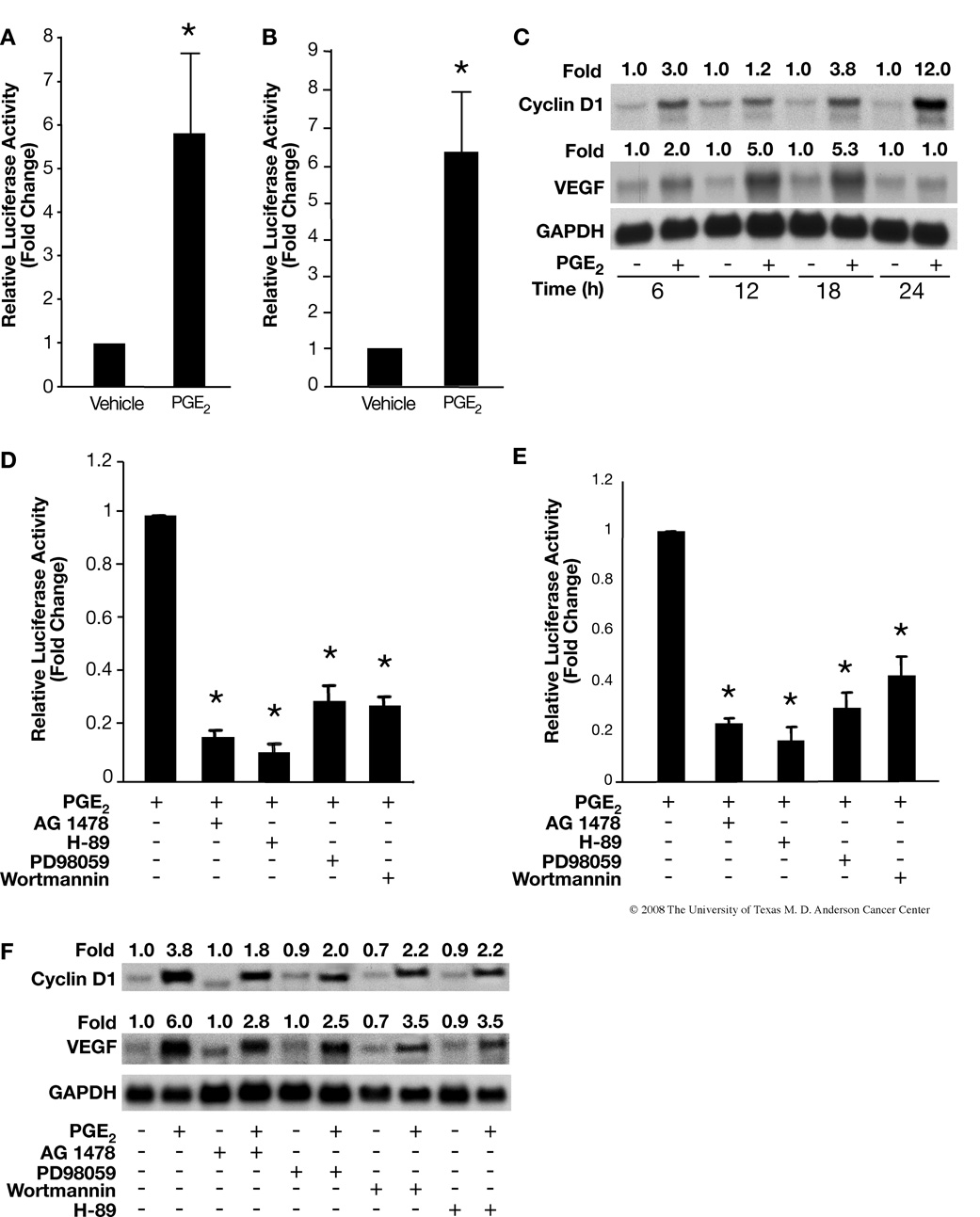
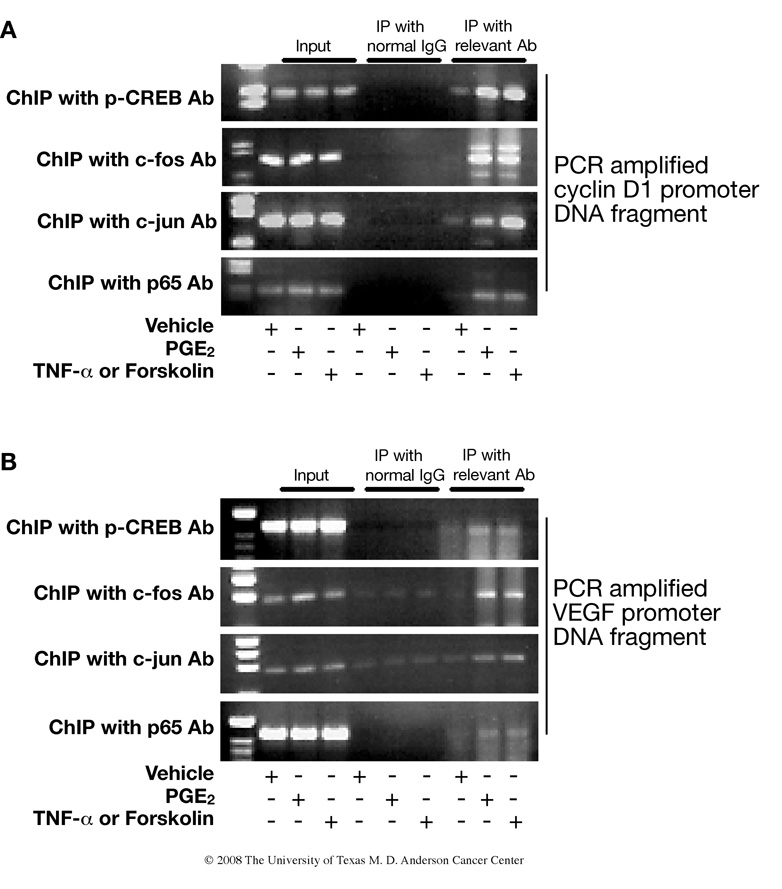
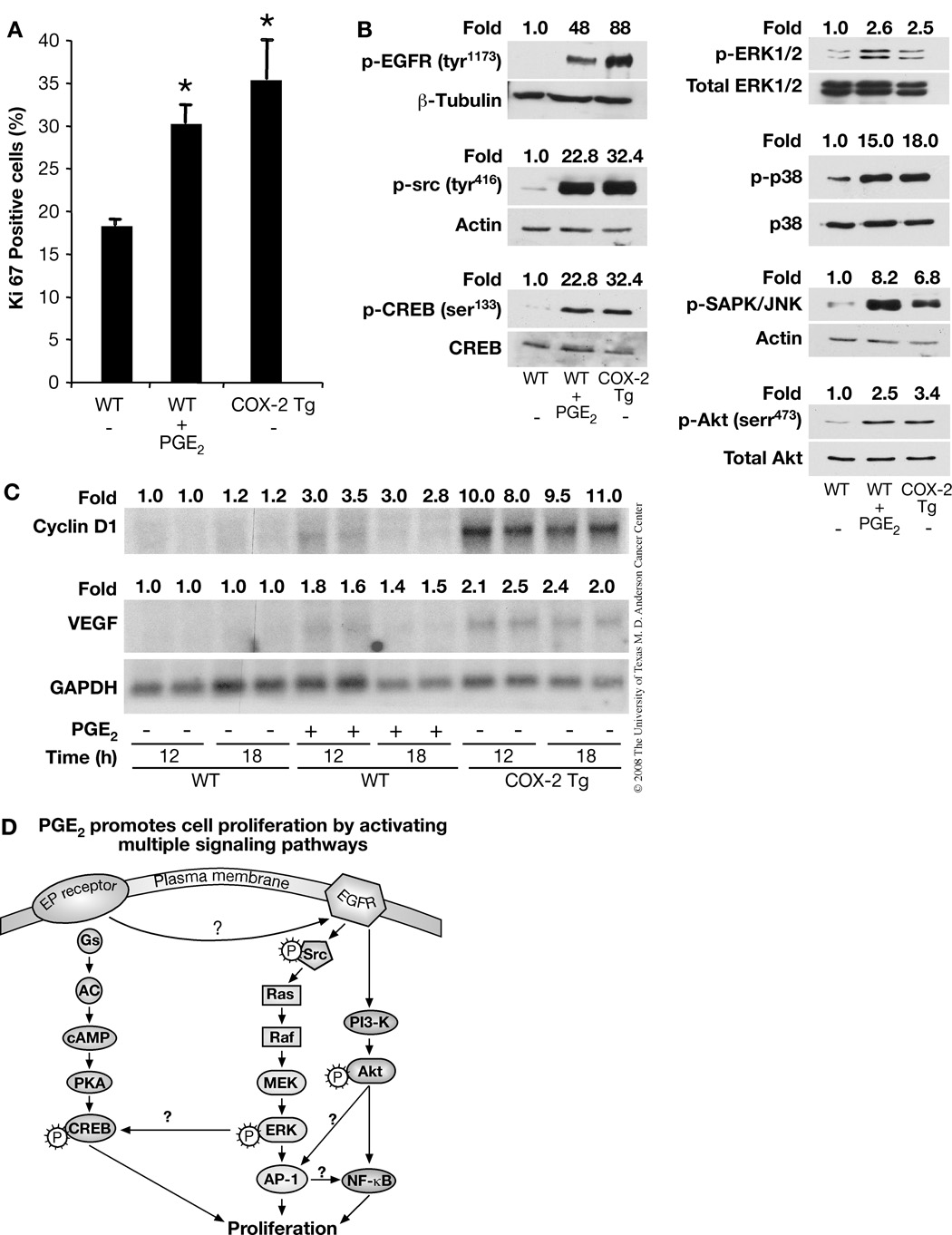
Although prostaglandin E2 (PGE2) has been shown by pharmacologic and genetic studies to be important in skin cancer, the molecular mechanism(s) by which it contributes to tumor growth is not well understood. In this study, we investigated the mechanisms by which PGE2 stimulates murine keratinocyte proliferation using in vitro and in vivo models. In primary mouse keratinocyte cultures, PGE2 activated the epidermal growth factor receptor (EGFR) and its downstream signaling pathways as well as increased cyclic AMP (cAMP) production and activated the cAMP response element binding protein (CREB). EGFR activation was not significantly inhibited by pretreatment with a c-src inhibitor (PP2), nor by a protein kinase A inhibitor (H-89). However, PGE2-stimulated extracellularly regulated kinase 1/2 (ERK1/2) activation was completely blocked by EGFR, ERK1/2, and phosphatidylinositol 3-kinase (PI3K) pathway inhibitors. In addition, these inhibitors attenuated the PGE2-induced proliferation, nuclear factor-kappa B, activator protein-1 (AP-1), and CREB binding to the promoter regions of the cyclin D1 and vascular endothelial growth factor (VEGF) genes and expression of cyclin D1 and VEGF in primary mouse keratinocytes. Similarly, in vivo, we found that WT mice treated with PGE2 and untreated cyclooxygenase-2-overexpressing transgenic mice had higher levels of cell proliferation and expression of cyclin D1 and VEGF, as well as higher levels of activated EGFR, nuclear factor-kappa B, AP-1, and CREB, than vehicle-treated WT mice. Our findings provide evidence for a link between cyclooxygenase-2 overexpression and EGFR-, ERK-, PI3K-, cAMP-mediated cell proliferation, and the tumor-promoting activity of PGE2 in mouse skin.
Figures
References
-
- Fischer SM. Eicosanoids and tumor promotion. In: Muhktar H, editor. Skin Cancer: Mechanisms and Human Relevance. Boca Raton, FL: CRC Press; 1995. pp. 129–144.
-
- Fischer SM. Prostaglandins and cancer. Front Biosci. 1997;2:d482–d500. - PubMed
-
- Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;23:144–150. - PubMed
-
- Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
