

Nitric oxide as inflammatory mediator in post-traumatic stress disorder (PTSD): evidence from an animal model
- PMID: 18568056
- PMCID: PMC2413191
- DOI: 10.2147/nedt.1.2.109.61049
Nitric oxide as inflammatory mediator in post-traumatic stress disorder (PTSD): evidence from an animal model
Abstract
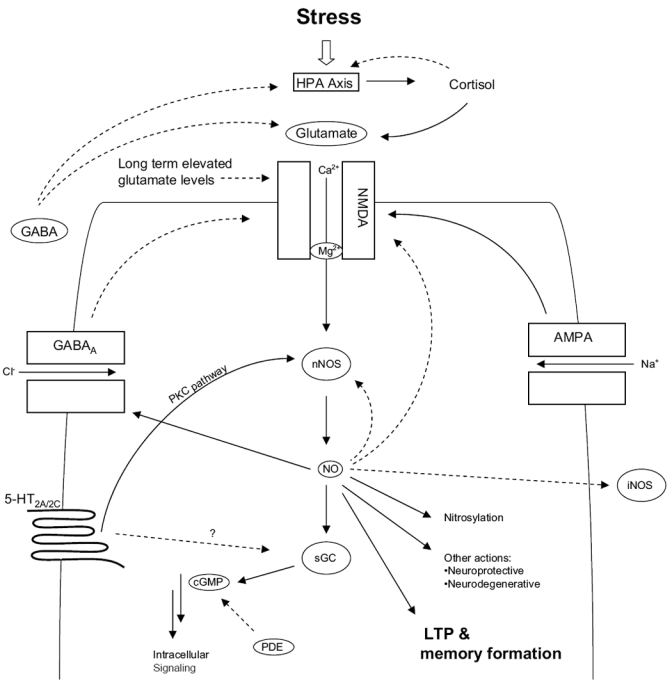
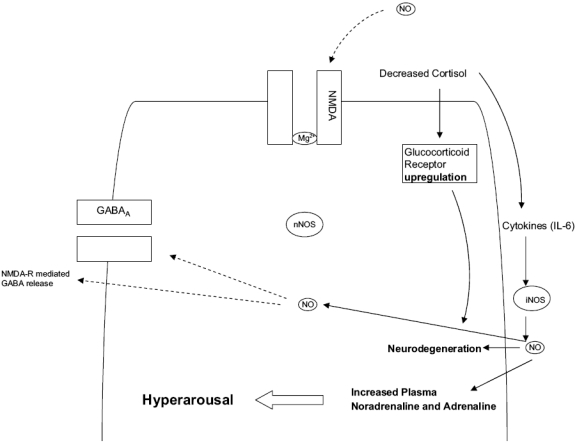
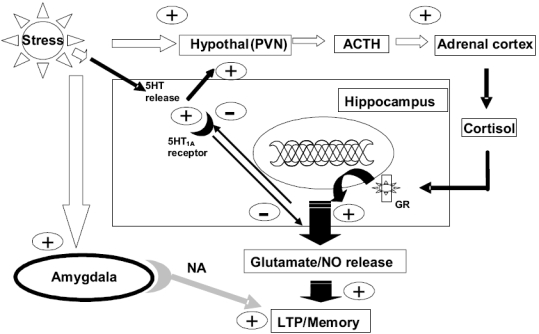
Post-traumatic stress disorder (PTSD) is a severe anxiety disorder that may develop after experiencing or witnessing a traumatic event. Recent clinical evidence has suggested the involvement of neurodegenerative pathology in the illness, particularly with brain imaging studies revealing a marked reduction in hippocampal volume. Of greater significance is that these anatomical changes appear to be positively correlated with the degree of cognitive deficit noted in these patients. Stress-induced increases in plasma cortisol have been implicated in this apparent atrophy. Although not definitive, clinical studies have observed a marked suppression of plasma cortisol in PTSD. The basis for hippocampal neurodegeneration and cognitive decline therefore remains unclear. Stress and glucocorticoids increase glutamate release, which is recognized as an important mediator of glucocorticoid-induced neurotoxicity. Recent preclinical studies have also noted that glutamate and nitric oxide (NO) play a causal role in anxiety-related behaviors. Because of the prominent role of NO in neuronal toxicity, cellular memory processes, and as a neuromodulator, nitrergic pathways may have an important role in stress-related hippocampal degenerative pathology and cognitive deficits seen in patients with PTSD. This paper reviews the preclinical evidence for involvement of the NO-pathway in PTSD, and emphasizes studies that have addressed these issues using time-dependent sensitization - a putative animal model of PTSD.
Keywords: GABA; NOS; PTSD; glucocorticoids; glutamate; nitric oxide; stress; time-dependent sensitization (TDS).
Figures
Similar articles
-
Stress-restress evokes sustained iNOS activity and altered GABA levels and NMDA receptors in rat hippocampus.Psychopharmacology (Berl). 2004 Oct;175(4):494-502. doi: 10.1007/s00213-004-1836-4. Psychopharmacology (Berl). 2004. PMID: 15138761
-
Endocrine, cognitive and hippocampal/cortical 5HT 1A/2A receptor changes evoked by a time-dependent sensitisation (TDS) stress model in rats.Brain Res. 2003 Sep 5;983(1-2):97-107. doi: 10.1016/s0006-8993(03)03033-6. Brain Res. 2003. PMID: 12914970
-
[Posttraumatic stress disorder (PTSD) as a consequence of the interaction between an individual genetic susceptibility, a traumatogenic event and a social context].Encephale. 2012 Oct;38(5):373-80. doi: 10.1016/j.encep.2011.12.003. Epub 2012 Jan 24. Encephale. 2012. PMID: 23062450 Review. French.
-
The role of glucocorticoids, catecholamines and endocannabinoids in the development of traumatic memories and posttraumatic stress symptoms in survivors of critical illness.Neurobiol Learn Mem. 2014 Jul;112:68-74. doi: 10.1016/j.nlm.2013.10.003. Epub 2013 Oct 11. Neurobiol Learn Mem. 2014. PMID: 24125890 Review.
-
Psychobiology of the acute stress response and its relationship to the psychobiology of post-traumatic stress disorder.Psychiatr Clin North Am. 2002 Jun;25(2):385-95. doi: 10.1016/s0193-953x(01)00005-3. Psychiatr Clin North Am. 2002. PMID: 12136506 Review.
Cited by
-
A Review of Biomarkers in Mood and Psychotic Disorders: A Dissection of Clinical vs. Preclinical Correlates.Curr Neuropharmacol. 2015;13(3):324-68. doi: 10.2174/1570159x13666150307004545. Curr Neuropharmacol. 2015. PMID: 26411964 Free PMC article. Review.
-
Emotional processes in patients undergoing coronary artery bypass graft surgeries with extracorporeal circulation in view of selected indicators of the inflammatory condition.Med Sci Monit. 2015 Jan 9;21:105-17. doi: 10.12659/MSM.892372. Med Sci Monit. 2015. PMID: 25573296 Free PMC article.
-
Autonomic and inflammatory consequences of posttraumatic stress disorder and the link to cardiovascular disease.Am J Physiol Regul Integr Comp Physiol. 2015 Aug 15;309(4):R315-21. doi: 10.1152/ajpregu.00343.2014. Epub 2015 Jun 10. Am J Physiol Regul Integr Comp Physiol. 2015. PMID: 26062635 Free PMC article. Review.
-
Oxidative Dysregulation in Early Life Stress and Posttraumatic Stress Disorder: A Comprehensive Review.Brain Sci. 2021 May 29;11(6):723. doi: 10.3390/brainsci11060723. Brain Sci. 2021. PMID: 34072322 Free PMC article. Review.
-
Brain vitamin D3-auto/paracrine system in relation to structural, neurophysiological, and behavioral disturbances associated with glucocorticoid-induced neurotoxicity.Front Cell Neurosci. 2023 Mar 20;17:1133400. doi: 10.3389/fncel.2023.1133400. eCollection 2023. Front Cell Neurosci. 2023. PMID: 37020845 Free PMC article.
References
-
- Adler A. Neuropsychiatric complications in victims of Boston’s Coconut Grove disaster. J Am Med Assoc. 1993;123:1098–101.
-
- Agrypolous SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs. Part 2: Pharmacological treatments of anxiety. Pharmacol Therap. 2000;88:213–27. - PubMed
-
- Akirav I, Sandi C, Richter-Levin G. Differential activation of hippocampus and amygdala following spatial learning under stress. Eur J Neurosci. 2001;14:719–25. - PubMed
-
- [APA] American Psychiatric Association. Diagnostic and statistical manual of mental disorders (DSM-IV) 4. Washington: APA; 1994.
-
- Baba H, Suzuki T, Arai H, et al. Expression of nNOS and soluble guanylate cyclase in schizophrenic brain. Neuroreport. 2004;15:677–80. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
