

Case Reports
doi: 10.1016/j.ajhg.2008.05.015.
Epub 2008 Jun 19.
FOXG1 is responsible for the congenital variant of Rett syndrome
Affiliations
- PMID: 18571142
- PMCID: PMC2443837
- DOI: 10.1016/j.ajhg.2008.05.015
Item in Clipboard
Case Reports
FOXG1 is responsible for the congenital variant of Rett syndrome
Am J Hum Genet.
2008 Jul.
Abstract
Rett syndrome is a severe neurodevelopmental disease caused by mutations in the X-linked gene encoding for the methyl-CpG-binding protein MeCP2. Here, we report the identification of FOXG1-truncating mutations in two patients affected by the congenital variant of Rett syndrome. FOXG1 encodes a brain-specific transcriptional repressor that is essential for early development of the telencephalon. Molecular analysis revealed that Foxg1 might also share common molecular mechanisms with MeCP2 during neuronal development, exhibiting partially overlapping expression domain in postnatal cortex and neuronal subnuclear localization.
Figures
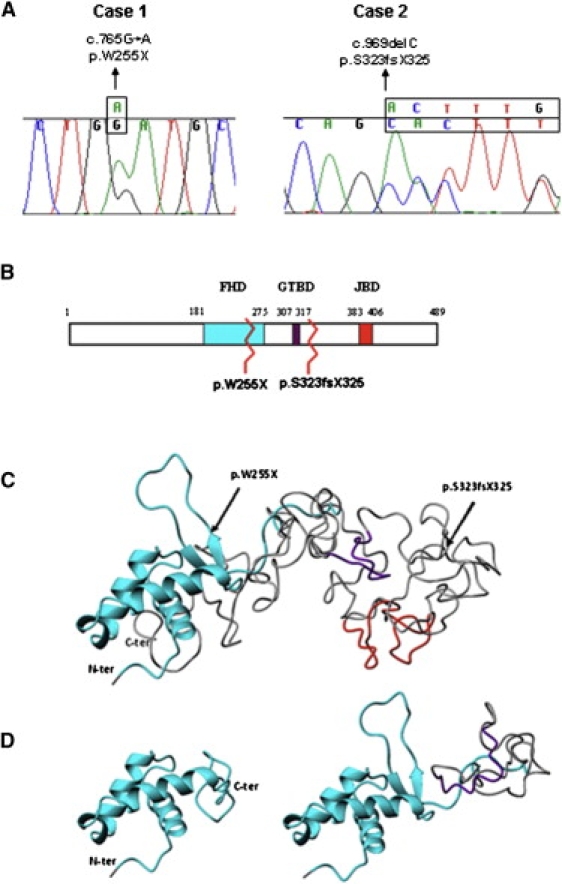
FOXG1 Mutations and Alterations of the Functional Domains (A) Sequence tracing of FOXG1 mutations in the two patients. Mutated bases are indicated above the line. (B) Schematic representation of FoxG1 protein. The three main functional domains are shown: the DNA binding fork-head domain in light blue (FHD), the Groucho-binding domain in violet (GTBD), and the JARID1B binding domain in red (JBD). The numbers at the top refer to the amino acid positions. Mutations are indicated by zigzag lines. (C and D) Ribbon representation of the tertiary structure obtained with Phyre v.0.2 software. (C) shows the structure of the region containing the three functional domains of wild-type protein (amino acids 180–489). Arrows highlight the two mutations. The FHD domain (cyan) consists of three alpha helices and one beta hairpin (two beta strands and one loop), whereas the GTBD (violet) and JBD (red) domains are random coiled. (D) shows structural modification after p.W255X (left) and p.S323fsX325 (right) mutations. The p.W255X mutation determines a protein truncation just after the second beta strand leading to the loss of the beta hairpin and thus preventing DNA binding. The p.S323fsX325 mutation leaves the FHD domain intact and truncates the protein just after GTBD, inducing conformational changes that lead to its misfolding.

Pictures of the Two Congenital RTT Patients Case 1 (#156) is shown on the left; case 2 (#868) is shown on the right. They show peculiar jerky movements of the upper limbs frequently pushed in different directions accompanied by continuous flexion-extension, wringing movements of the fingers of the hands. The hands were brought together in hand-washing and hand-mouth stereotypic activities, which were intense and present all the time they were awake. Similar flexion-extension movements of the toes were noticed in the feet. The double scoliosis of case 1 is clearly evident, whereas the other girl maintained a straight vertebral column as often occurs in RTT in the first decade. Teeth grinding was present, and the tongue often protruded out from the mouth.
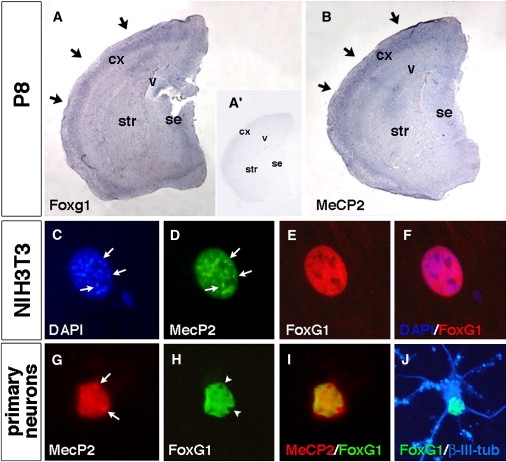
FoxG1 and MeCP2 Expression Domain in Postnatal Cortex and Neuronal Subnuclear Localization (A and B) Expression analysis by in situ hybridization of Foxg1 and MeCP2 on P8 postnatal forebrain tissue. As shown in (A), Foxg1 expression is found in differentiating and mature cortical neurons in the definitive cortical plate (indicated by arrows in [A]) similar to the MeCP2 expression pattern (indicated by arrows in [B]). In (A′), the inset shows background staining with a sense cRNA for Foxg1 in the same in situ hybridization conditions used for (A) and (B). (C–J) FoxG1 and MeCP2 sub-cellular localization in non-neural and primary neurons. As shown in (C)–(F), in NIH 3T3 cells, MeCP2-GFP exogenous protein has a diffuse nuclear localization with accumulation in the heterochromatic foci (indicated by arrows in [D]) as identified when compared with DAPI staining (indicated by arrows in [C]). (E) and (F) show that conversely, FoxG1-flag exogenous protein displays a widespread nuclear localization without enrichment in heterochromatic sites. (G)–(J) show FoxG1 and MeCP2 localization in 12DIV (days in vitro) primary hippocampal neurons. In (G), MeCP2 endogenous protein is accumulated in heterochromatic foci (indicated by arrows). As shown in (H), FoxG1-flag exogenous nuclear localization is excluded from heterochromatic puncta (indicated by arrowheads). As shown in (I), MeCP2 and FoxG1-flag colocalize in the nuclear compartment outside the heterochromatic foci. As shown in (J), Nuclear FoxG1-flag localization is detected in a differentiated β-III-tubulin-positive neuron. The following abbreviations are used: cx, cerebral cortex; se, septum; str, striatum; and v, ventricle.
References
-
- Chahrour M., Zoghbi H.Y. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. - PubMed
-
- Hagberg B.A., Skjeldal O.H. Rett variants: A suggested model for inclusion criteria. Pediatr. Neurol. 1994;11:5–11. - PubMed
-
- De Bona C., Zappella M., Hayek G., Meloni I., Vitelli F., Bruttini M., Cusano R., Loffredo P., Longo I., Renieri A. Preserved speech variant is allelic of classic Rett syndrome. Eur. J. Hum. Genet. 2000;8:325–330. - PubMed
-
- Tao J., Van Esch H., Hagedorn-Greiwe M., Hoffmann K., Moser B., Raynaud M., Sperner J., Fryns J., Schwinger E., Gecz J. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am. J. Hum. Genet. 2004;75:1149–1154. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
