

Natural selection on genes that underlie human disease susceptibility
- PMID: 18571414
- PMCID: PMC2474766
- DOI: 10.1016/j.cub.2008.04.074
Natural selection on genes that underlie human disease susceptibility
Abstract
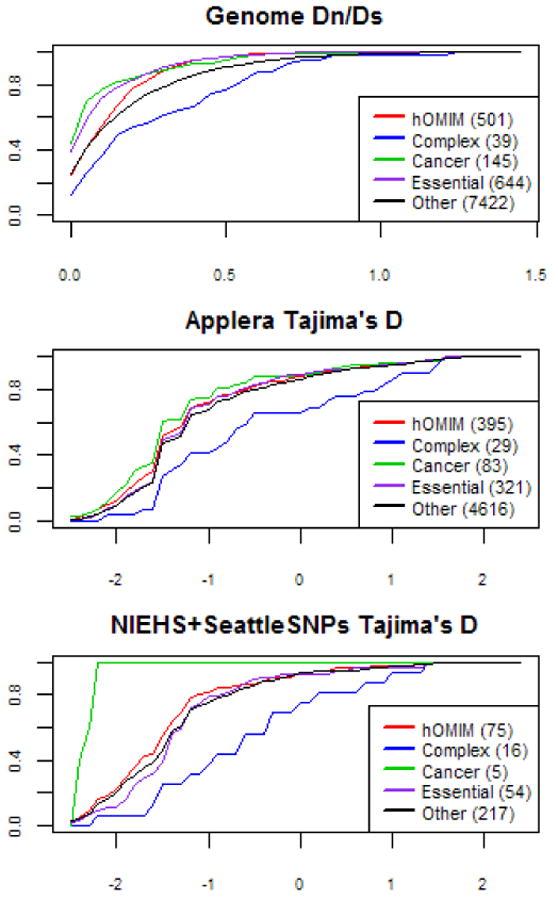
What evolutionary forces shape genes that contribute to the risk of human disease? Do similar selective pressures act on alleles that underlie simple versus complex disorders [1-3]? Answers to these questions will shed light onto the origin of human disorders (e.g., [4]) and help to predict the population frequencies of alleles that contribute to disease risk, with important implications for the efficient design of mapping studies [5-7]. As a first step toward addressing these questions, we created a hand-curated version of the Mendelian Inheritance in Man database (OMIM). We then examined selective pressures on Mendelian-disease genes, genes that contribute to complex-disease risk, and genes known to be essential in mouse by analyzing patterns of human polymorphism and of divergence between human and rhesus macaque. We found that Mendelian-disease genes appear to be under widespread purifying selection, especially when the disease mutations are dominant (rather than recessive). In contrast, the class of genes that influence complex-disease risk shows little signs of evolutionary conservation, possibly because this category includes targets of both purifying and positive selection.
Figures
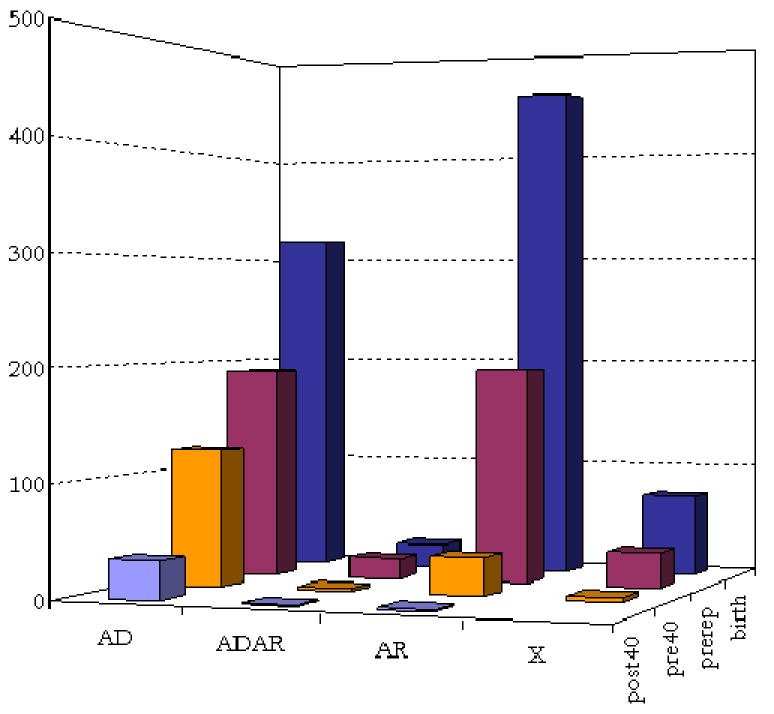
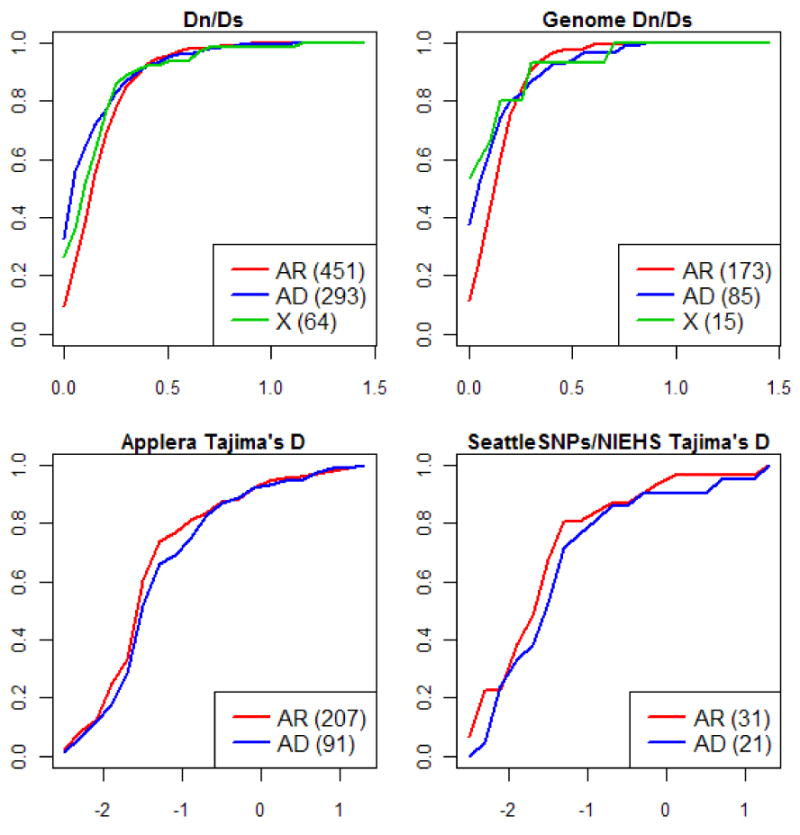
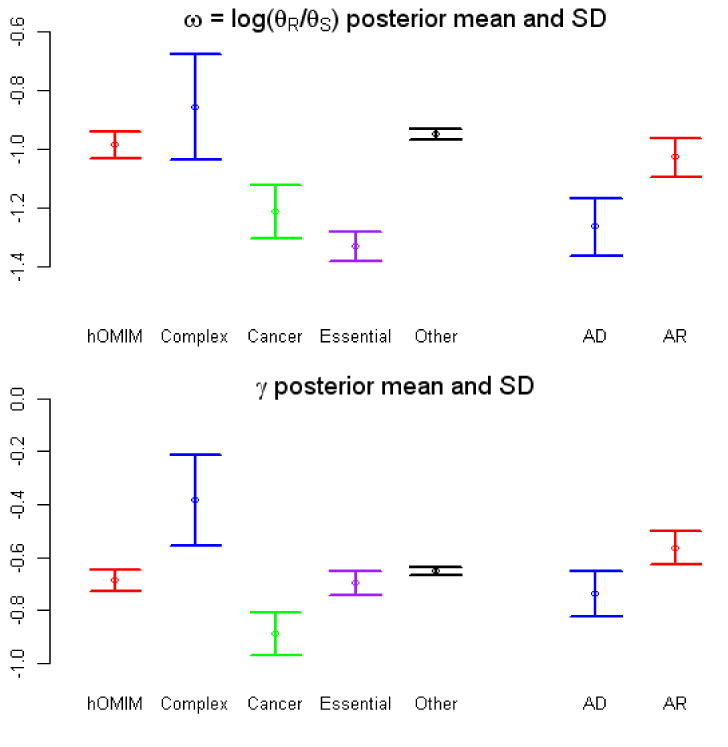
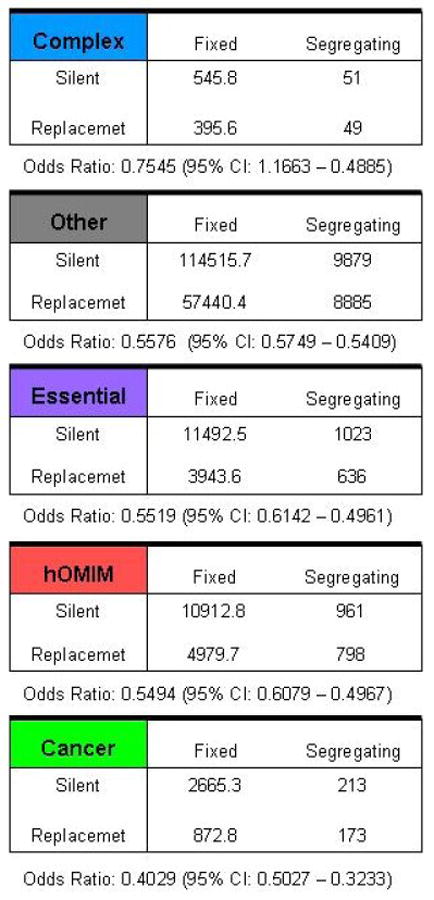
References
-
- Zwick ME, Cutler DJ, Chakravarti A. Patterns of genetic variation in Mendelian and complex traits. Annu Rev Genomics Hum Genet. 2000;1:387–407. - PubMed
-
- Di Rienzo A. Population genetics models of common diseases. Curr Opin Genet Dev. 2006;16:630–636. - PubMed
-
- Keller MC, Miller G. Resolving the paradox of common, harmful, heritable mental disorders: which evolutionary genetic models work best? Behav Brain Sci. 2006;29:385–404. discussion 405-352. - PubMed
-
- Pritchard JK, Cox NJ. The allelic architecture of human disease genes: common disease-common variant…or not? Hum Mol Genet. 2002;11:2417–2423. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
