

A novel human disease with abnormal prion protein sensitive to protease
- PMID: 18571782
- PMCID: PMC2767200
- DOI: 10.1002/ana.21420
A novel human disease with abnormal prion protein sensitive to protease
Abstract
Objective: To report a novel prion disease characterized by distinct histopathological and immunostaining features, and associated with an abnormal isoform of the prion protein (PrP) that, contrary to the common prion diseases, is predominantly sensitive to protease digestion.
Methods: Eleven subjects were investigated at the National Prion Disease Pathology Surveillance Center for clinical, histopathological, immunohistochemical, genotypical, and PrP characteristics.
Results: Patients presented with behavioral and psychiatric manifestations on average at 62 years, whereas mean disease duration was 20 months. The type of spongiform degeneration, the PrP immunostaining pattern, and the presence of microplaques distinguished these cases from those with known prion diseases. Typical protease-resistant PrP was undetectable in the cerebral neocortex with standard diagnostic procedures. After enrichment, abnormal PrP was detected at concentrations 16 times lower than common prion diseases; it included nearly 4 times less protease-resistant PrP, which formed a distinct electrophoretic profile. The subjects examined comprised about 3% of sporadic cases evaluated by the National Prion Disease Pathology Surveillance Center. Although several subjects had family histories of dementia, no mutations were found in the PrP gene open reading frame.
Interpretation: The distinct histopathological, PrP immunohistochemical, and physicochemical features, together with the homogeneous genotype, indicate that this is a previously unidentified type of disease involving the PrP, which we designated "protease-sensitive prionopathy" (or PSPr). Protease-sensitive prionopathy is not rare among prion diseases, and it may be even more prevalent than our data indicate because protease-sensitive prionopathy cases are likely also to be classified within the group of non-Alzheimer's dementias.
Figures
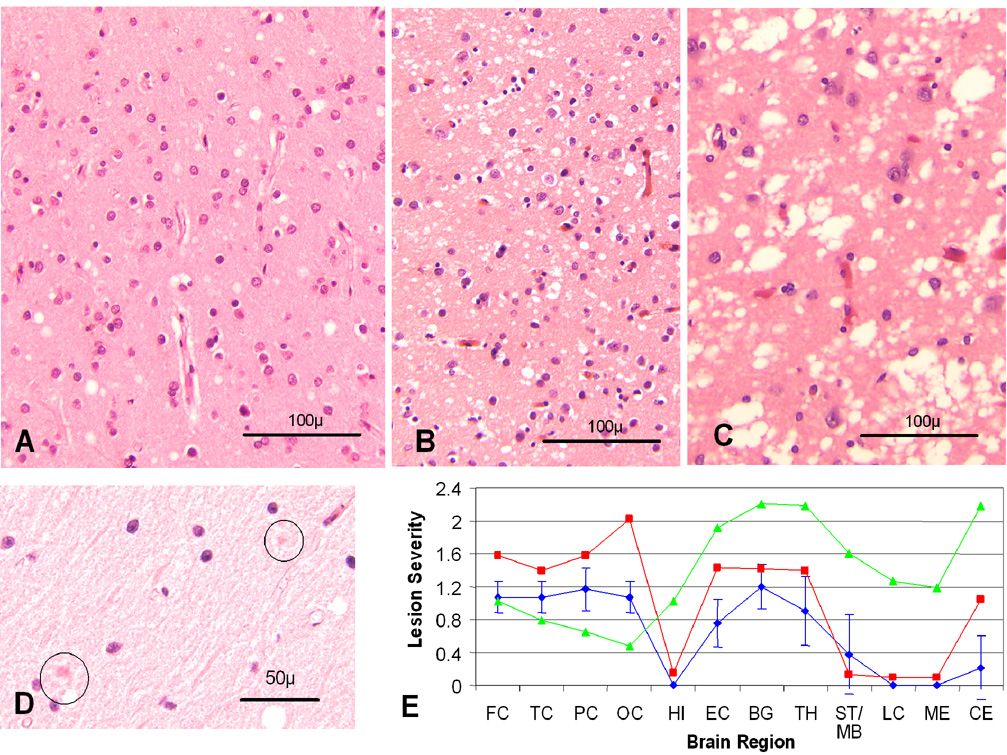
 ), sCJDMM1 (
), sCJDMM1 ( ) and sCJDVV2 (
) and sCJDVV2 ( ); (FC, TC, PC and OC: frontal, temporal, parietal and occipital cortices; HI: CA1 of hippocampus; EC: entorhinal cortex; BG: basal ganglia; TH: thalamus medial-dorsal nucleus; MB/ST: mid brain in PSPr, substantia nigra in sCJDMM1 and sCJDVV2; LC: pons; ME: medulla; CE: cerebellar cortex). The vertical bars refer to standard deviations. In sCJDMM1 and sCJDVV2, for which data were adapted from Parchi et al., standard deviations were omitted for clarity. Spongiform degeneration was scored on a 0 to 4 scale (0-not detectable, 1-mild, 2-moderate, 3-severe, and 4-confluent); astrogliosis on a 0 to 3 scale (0-not detectable, 1-mild, 2-moderate, and 3-severe).
); (FC, TC, PC and OC: frontal, temporal, parietal and occipital cortices; HI: CA1 of hippocampus; EC: entorhinal cortex; BG: basal ganglia; TH: thalamus medial-dorsal nucleus; MB/ST: mid brain in PSPr, substantia nigra in sCJDMM1 and sCJDVV2; LC: pons; ME: medulla; CE: cerebellar cortex). The vertical bars refer to standard deviations. In sCJDMM1 and sCJDVV2, for which data were adapted from Parchi et al., standard deviations were omitted for clarity. Spongiform degeneration was scored on a 0 to 4 scale (0-not detectable, 1-mild, 2-moderate, 3-severe, and 4-confluent); astrogliosis on a 0 to 3 scale (0-not detectable, 1-mild, 2-moderate, and 3-severe).



Comment in
-
A new prionopathy.Ann Neurol. 2008 Jun;63(6):677-8. doi: 10.1002/ana.21447. Ann Neurol. 2008. PMID: 18570344 No abstract available.
References
-
- Gambetti P, Kong Q, Zou WQ, et al. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–239. - PubMed
-
- Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. - PubMed
-
- Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–778. - PubMed
-
- Kong Q, Surewicz WK, Peterson RB, et al. Inherited prion diseases. In: Prusiner SB, editor. Prion Biology and Disease. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory press; 2004. pp. 673–775.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
