

The protein folding problem
- PMID: 18573083
- PMCID: PMC2443096
- DOI: 10.1146/annurev.biophys.37.092707.153558
The protein folding problem
Abstract
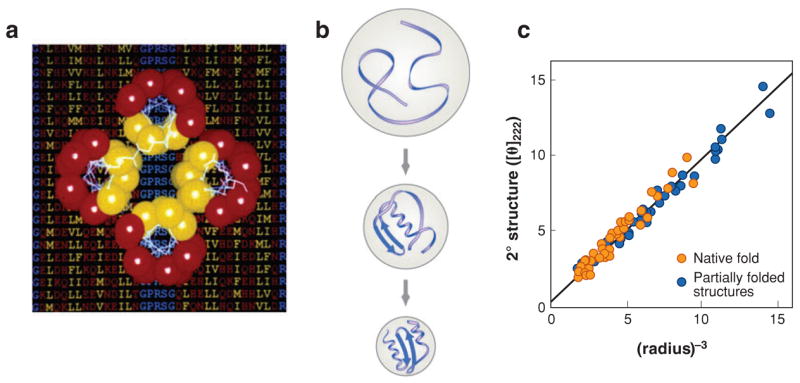
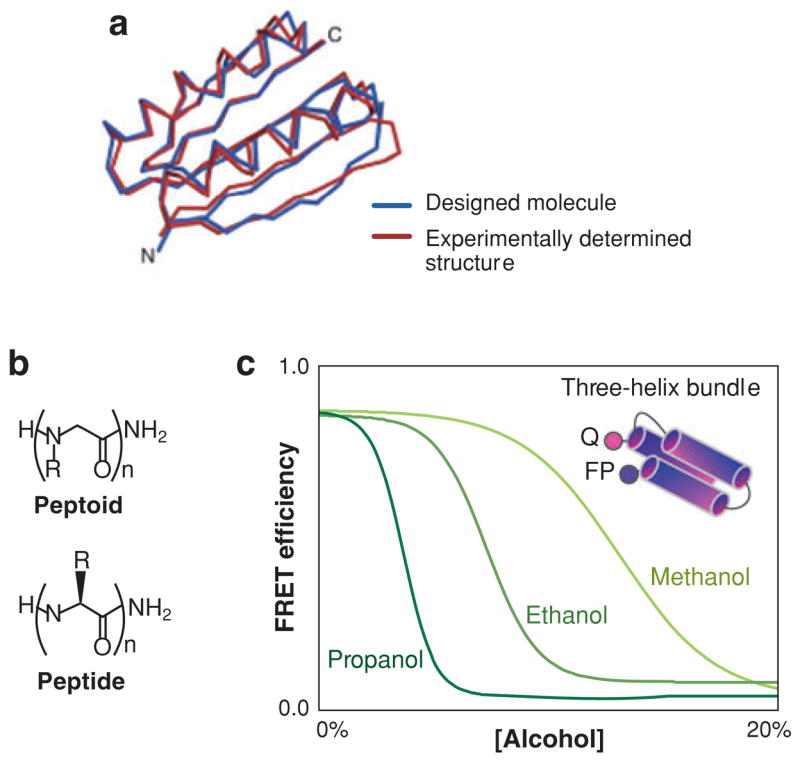
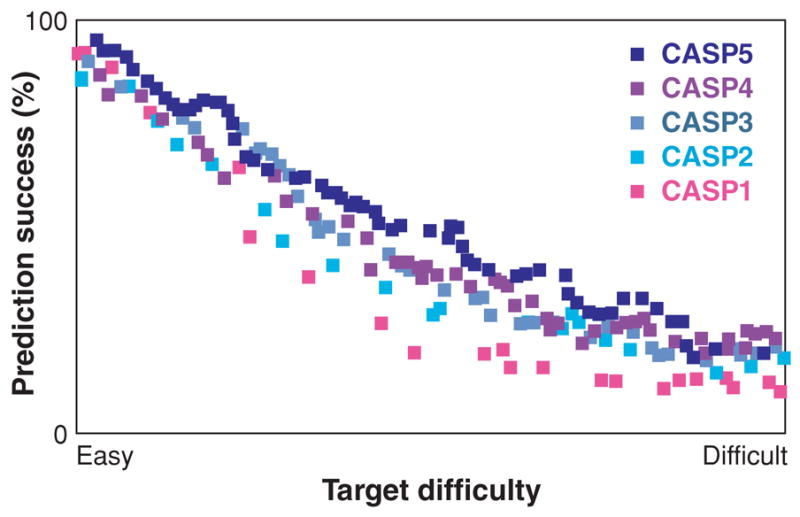
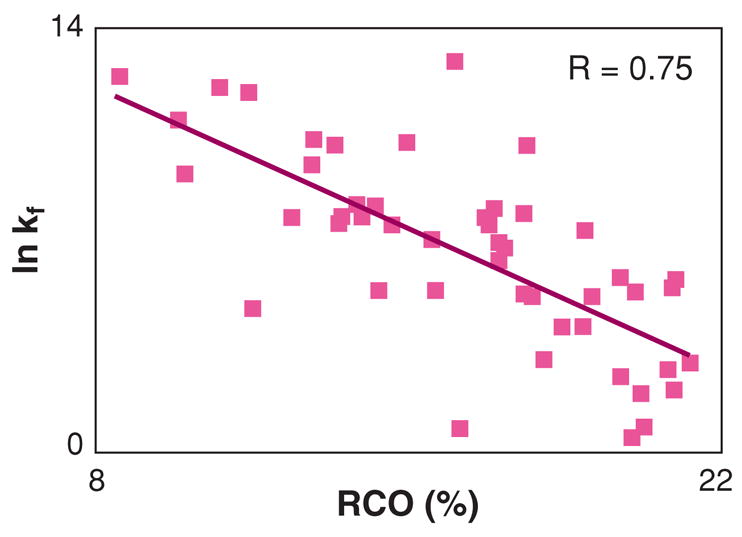
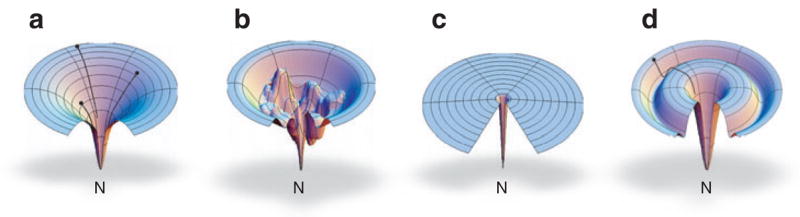
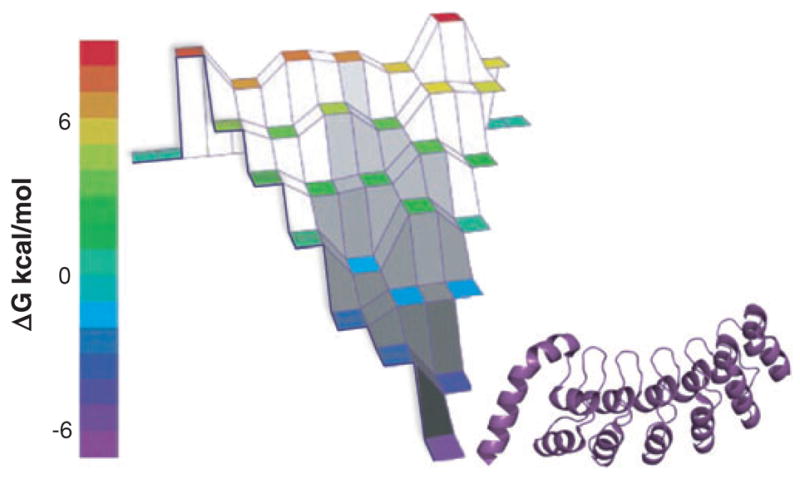
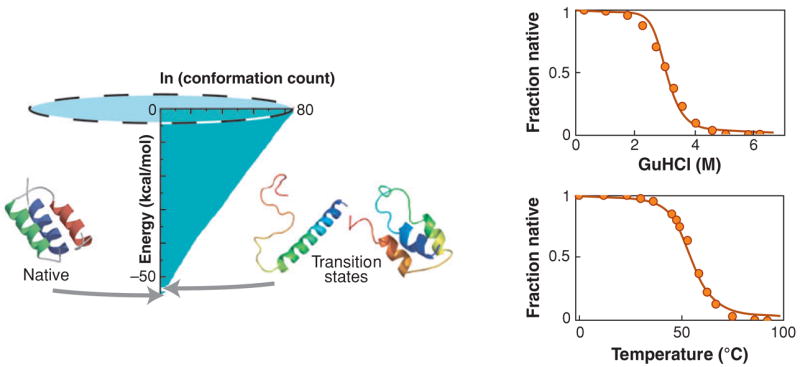
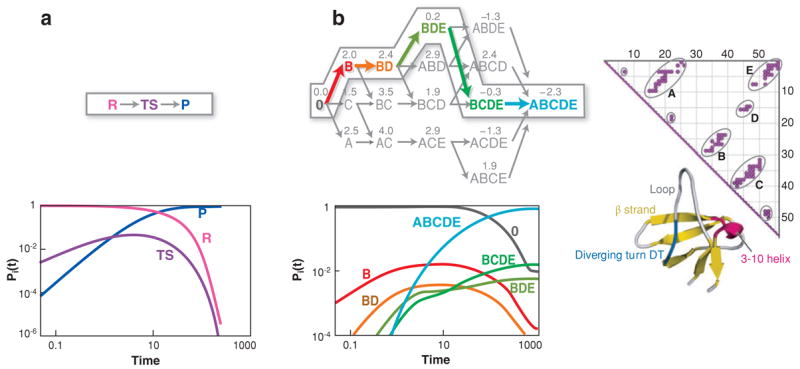
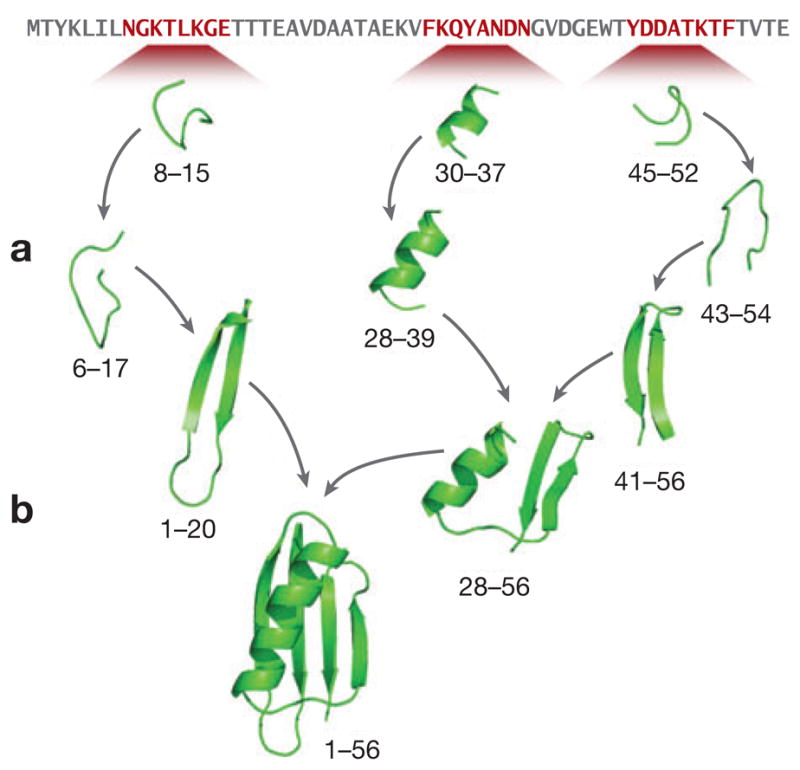
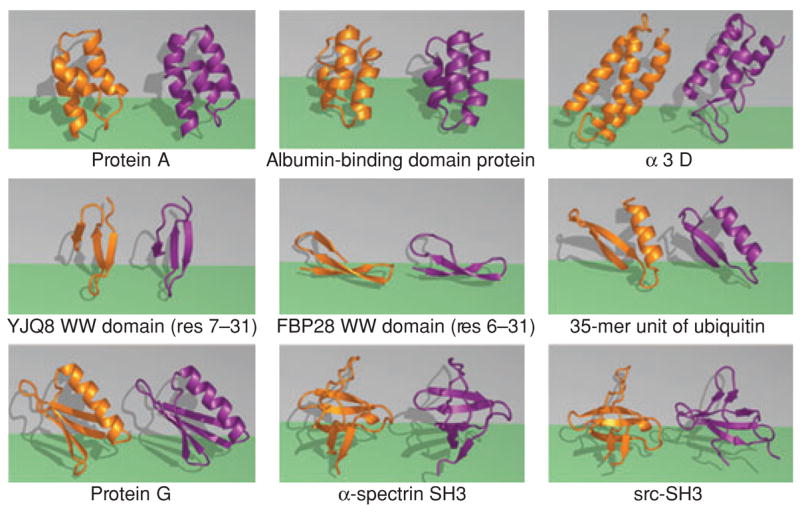
The "protein folding problem" consists of three closely related puzzles: (a) What is the folding code? (b) What is the folding mechanism? (c) Can we predict the native structure of a protein from its amino acid sequence? Once regarded as a grand challenge, protein folding has seen great progress in recent years. Now, foldable proteins and nonbiological polymers are being designed routinely and moving toward successful applications. The structures of small proteins are now often well predicted by computer methods. And, there is now a testable explanation for how a protein can fold so quickly: A protein solves its large global optimization problem as a series of smaller local optimization problems, growing and assembling the native structure from peptide fragments, local structures first.
Figures
References
-
- So much more to know. . . Science. 2005;309:78–102. - PubMed
-
- Allen F, Coteus P, Crumley P, Curioni A, Denneau M, et al. Blue gene: a vision for protein science using a petaflop supercomputer. IBM Syst J. 2001;40:310–27.
-
- Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–30. - PubMed
-
- Anfinsen CB, Scheraga HA. Experimental and theoretical aspects of protein folding. Adv Protein Chem. 1975;29:205–300. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
