

Activator protein 2alpha mediates parathyroid TGF-alpha self-induction in secondary hyperparathyroidism
- PMID: 18579641
- PMCID: PMC2551566
- DOI: 10.1681/ASN.2007111216
Activator protein 2alpha mediates parathyroid TGF-alpha self-induction in secondary hyperparathyroidism
Abstract
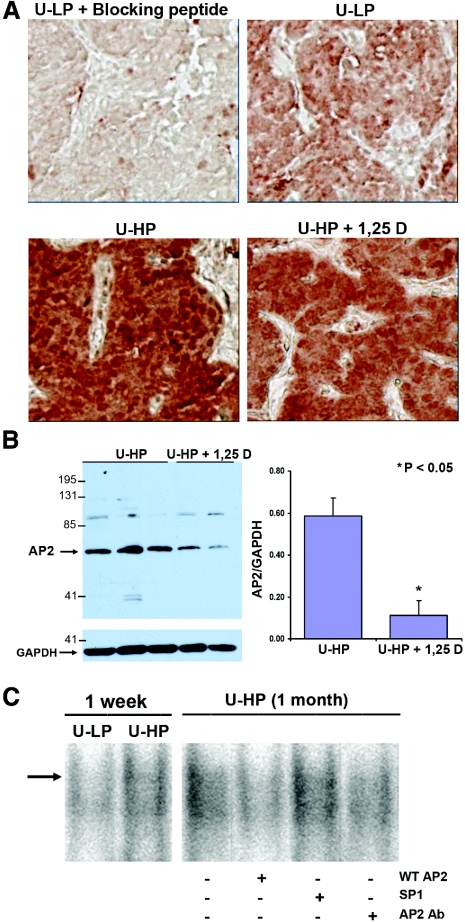
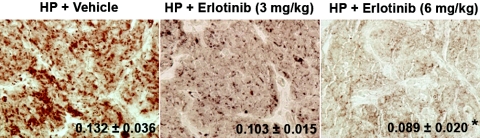
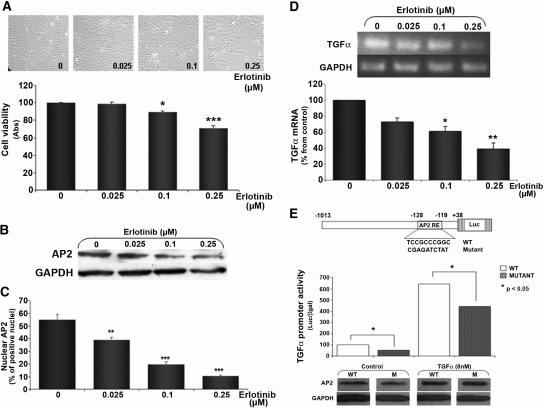
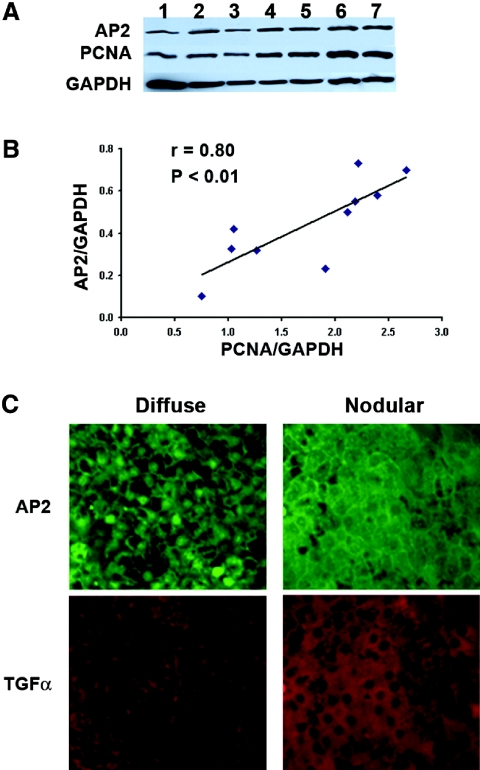
In secondary hyperparathyroidism, enhanced expression of TGF-alpha in the parathyroid leads to its own upregulation, generating a feed-forward loop for TGF-alpha activation of its receptor, EGFR receptor (EGFR), which promotes parathyroid hyperplasia. These studies examined the role of activator protein 2alpha (AP2), an inducer of TGF-alpha gene transcription, in the upregulation of parathyroid TGF-alpha in secondary hyperparathyroidism. In rat and human secondary hyperparathyroidism, parathyroid AP2 expression strongly correlated with TGF-alpha levels and with the rate of parathyroid growth, as expected. Furthermore, the increases in rat parathyroid content of AP2 and its binding to a consensus AP2 DNA sequence preceded the increase in TGF-alpha induced by high dietary phosphate. More significant, in A431 cells, which provide a model of enhanced TGF-alpha and TGF-alpha self-induction, mutating the core AP2 site of the human TGF-alpha promoter markedly impaired promoter activity induced by endogenous or exogenous TGF-alpha. Important for therapy, in five-sixths nephrectomized rats fed high-phosphate diets, inhibition of parathyroid TGF-alpha self-induction using erlotinib, a highly specific inhibitor of TGF-alpha/EGFR-driven signals, reduced AP2 expression dosage dependently. This suggests that the increases in parathyroid AP2 occur downstream of EGFR activation by TGF-alpha and are required for TGF-alpha self-induction. Indeed, in A431 cells, erlotinib inhibition of TGF-alpha self-induction caused parallel reductions in AP2 expression and nuclear localization, as well as TGF-alpha mRNA and protein levels. In summary, increased AP2 expression and transcriptional activity at the TGF-alpha promoter determine the severity of the hyperplasia driven by parathyroid TGF-alpha self-upregulation in secondary hyperparathyroidism.
Figures

References
-
- Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, Wang Y, Chung J, Emerick A, Greaser L, Elashoff RM, Salusky IB: Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med 342: 1478–1483, 2000 - PubMed
-
- Cozzolino M, Dusso AS, Slatopolsky E: Role of calcium-phosphate product and bone-associated proteins on vascular calcification in renal failure. J Am Soc Nephrol 12: 2511–2516, 2001 - PubMed
-
- Gonzalez EA, Martin KJ: Renal osteodystrophy: Pathogenesis and management. Nephrol Dial Transplant 10[Suppl 3]: 13–21, 1995 - PubMed
-
- Parfitt AM: The hyperparathyroidism of chronic renal failure: A disorder of growth. Kidney Int 52: 3–9, 1997 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
