

Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair
- PMID: 18584027
- PMCID: PMC2430616
- DOI: 10.1371/journal.pgen.1000110
Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair
Abstract
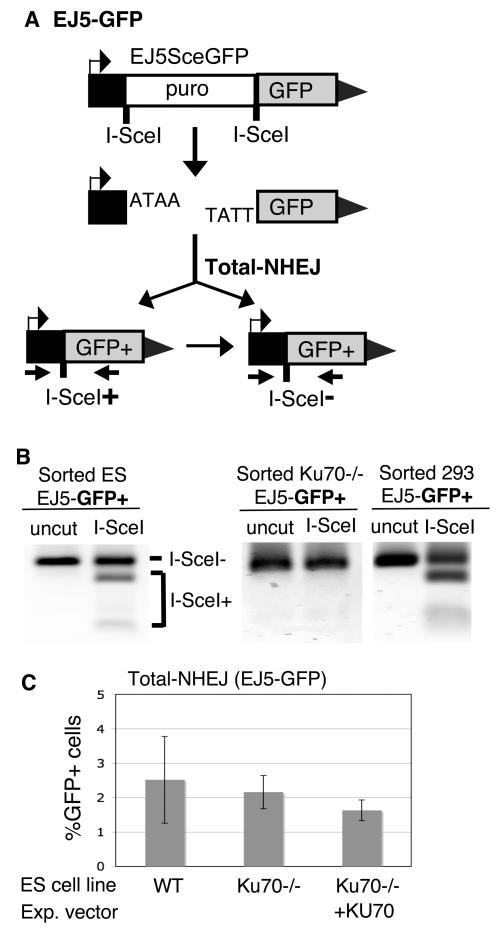
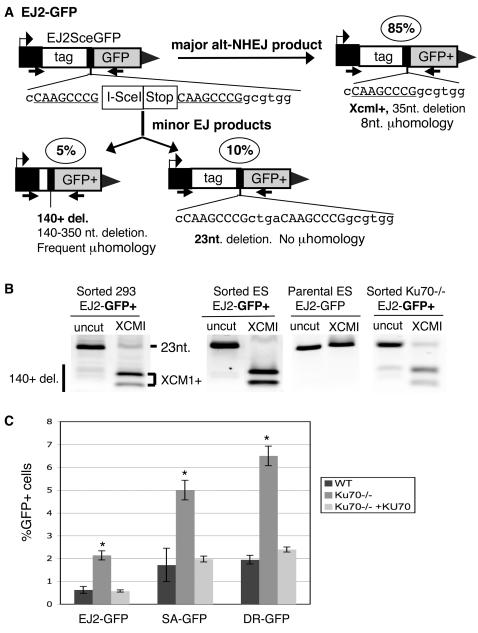
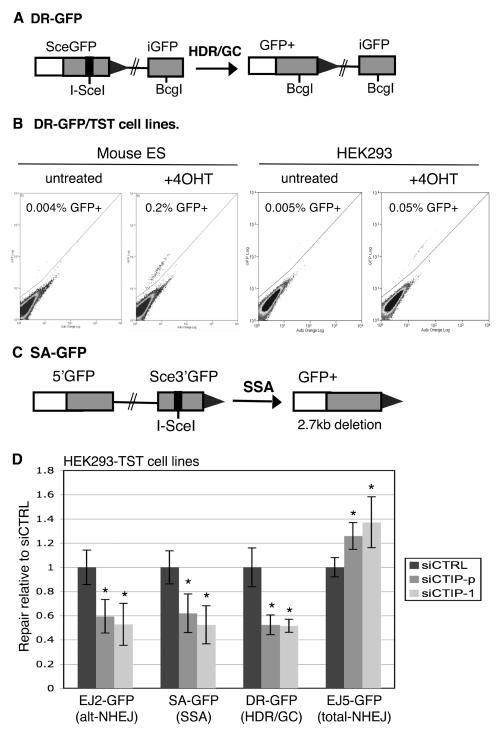
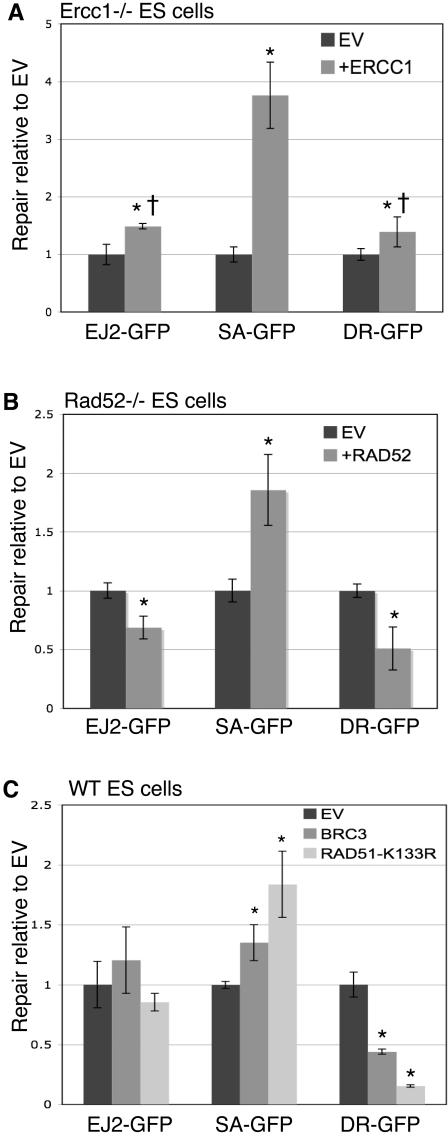
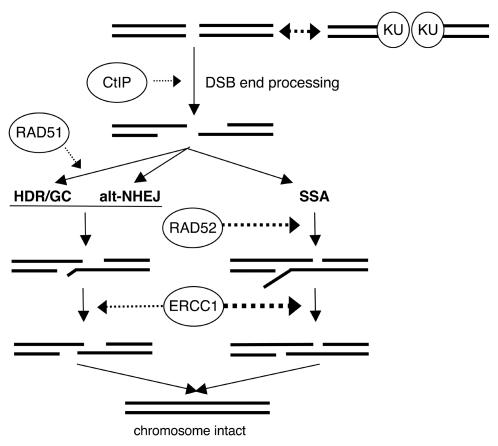
Characterizing the functional overlap and mutagenic potential of different pathways of chromosomal double-strand break (DSB) repair is important to understand how mutations arise during cancer development and treatment. To this end, we have compared the role of individual factors in three different pathways of mammalian DSB repair: alternative-nonhomologous end joining (alt-NHEJ), single-strand annealing (SSA), and homology directed repair (HDR/GC). Considering early steps of repair, we found that the DSB end-processing factors KU and CtIP affect all three pathways similarly, in that repair is suppressed by KU and promoted by CtIP. In contrast, both KU and CtIP appear dispensable for the absolute level of total-NHEJ between two tandem I-SceI-induced DSBs. During later steps of repair, we find that while the annealing and processing factors RAD52 and ERCC1 are important to promote SSA, both HDR/GC and alt-NHEJ are significantly less dependent upon these factors. As well, while disruption of RAD51 causes a decrease in HDR/GC and an increase in SSA, inhibition of this factor did not affect alt-NHEJ. These results suggest that the regulation of DSB end-processing via KU/CtIP is a common step during alt-NHEJ, SSA, and HDR/GC. However, at later steps of repair, alt-NHEJ is a mechanistically distinct pathway of DSB repair, and thus may play a unique role in mutagenesis during cancer development and therapy.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. - PubMed
-
- Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst) 2004;3:781–796. - PubMed
-
- Burma S, Chen BP, Chen DJ. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst) 2006;5:1042–1048. - PubMed
-
- Guirouilh-Barbat J, Huck S, Bertrand P, Pirzio L, Desmaze C, et al. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol Cell. 2004;14:611–623. - PubMed
-
- Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, et al. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109:811–821. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
