

No longer an innocent bystander: epithelial toll-like receptor signaling in the development of mucosal inflammation
- PMID: 18584047
- PMCID: PMC2435494
- DOI: 10.2119/2008-00035.Gribar
No longer an innocent bystander: epithelial toll-like receptor signaling in the development of mucosal inflammation
Abstract
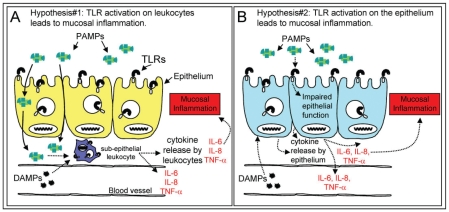
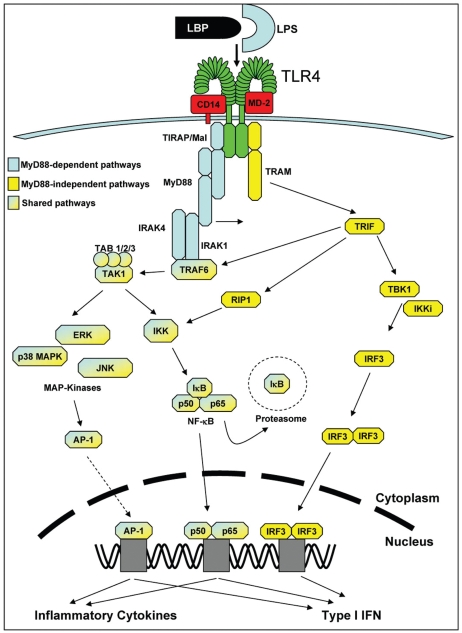
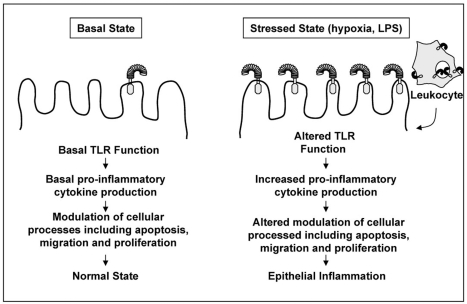
Diseases of mucosal inflammation represent important causes of morbidity and mortality, and have led to intense research efforts to understand the factors that lead to their development. It is well accepted that a breakdown of the normally impermeant epithelial barrier of the intestine, the lung, and the kidney is associated with the development of inflammatory disease in these organs, yet significant controversy exists as to how this breakdown actually occurs, and how such a breakdown may lead to inflammation. In this regard, much work has focused upon the role of the epithelium as an "innocent bystander," a target of a leukocyte-mediated inflammatory cascade that leads to its destruction in the mucosal inflammatory process. However, recent evidence from a variety of laboratories indicates that the epithelium is not merely a passive component in the steps that lead to mucosal inflammation, but is a central participant in the process. In addressing this controversy, we and others have determined that epithelial cells express Toll-like receptors (TLRs) of the innate immune system, and that activation of TLRs by endogenous and exogenous ligands may play a central role in determining the balance between a state of "mucosal homeostasis," as is required for optimal organ function, and "mucosal injury," leading to mucosal inflammation and barrier breakdown. In particular, activation of TLRs within intestinal epithelial cells leads to the development of cellular injury and impairment in mucosal repair in the pathogenesis of intestinal inflammation, while activation of TLRs in the lung and kidney may participate in the development of pneumonitis and nephritis respectively. Recent work in support of these concepts is extensively reviewed, while essential areas of further study that are required to determine the significance of epithelial TLR signaling during states of health and disease are outlined.
Figures
References
-
- Bibiloni R, Mangold M, Madsen KL, Fedorak RN, Tannock GW. The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn’s disease and ulcerative colitis patients. J Med Microbiol. 2006;55:1141–9. - PubMed
-
- Martinez-Medina M, Aldeguer X, Gonzalez-Huix F, Acero D, Garcia-Gil LJ. Abnormal microbiota composition in the ileocolonic mucosa of Crohn’s disease patients as revealed by polymerase chain reaction-denaturing gradient gel electrophoresis. Inflamm Bowel Dis. 2006;12:1136–45. - PubMed
-
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–34. - PubMed
-
- Anand R, Leaphart CL, Mollen K, Hackam D. The role of the intestinal barrier in the pathogenesis of necrotizing enterocolitis. Shock. 2007;27:124–33. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
