

Genetic basis of virulence attenuation revealed by comparative genomic analysis of Mycobacterium tuberculosis strain H37Ra versus H37Rv
- PMID: 18584054
- PMCID: PMC2440308
- DOI: 10.1371/journal.pone.0002375
Genetic basis of virulence attenuation revealed by comparative genomic analysis of Mycobacterium tuberculosis strain H37Ra versus H37Rv
Abstract
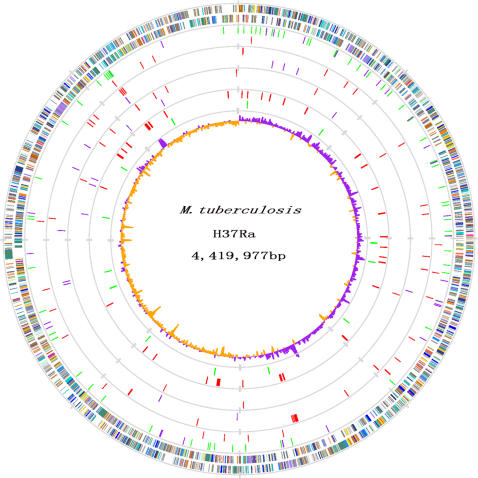
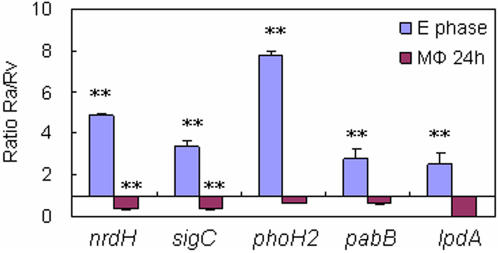
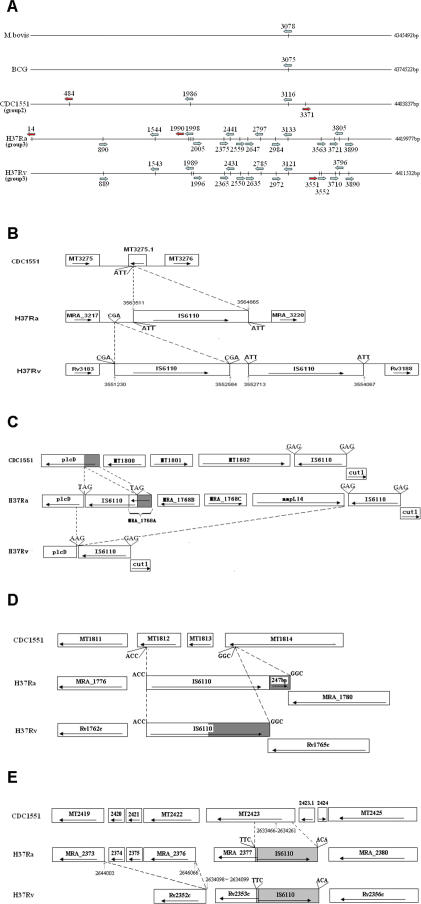
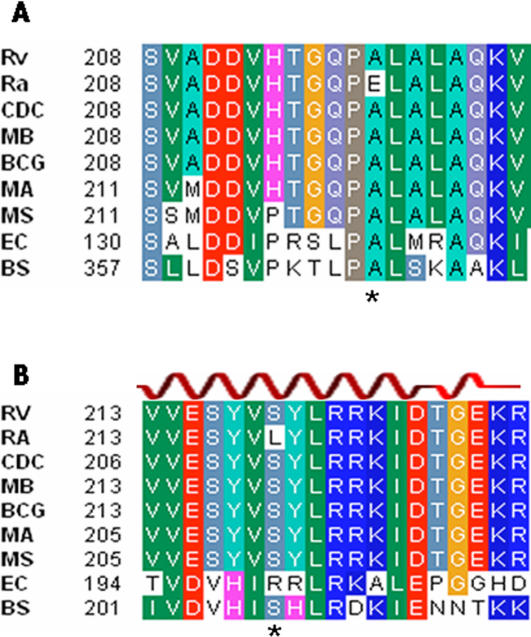
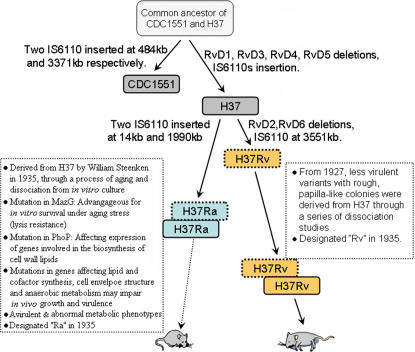
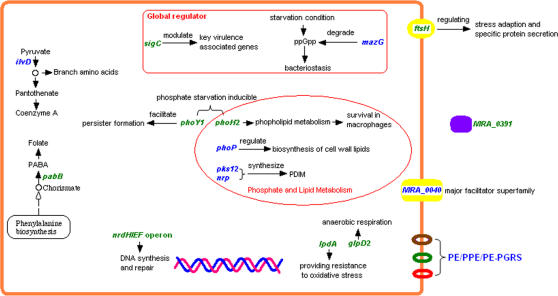
Tuberculosis, caused by Mycobacterium tuberculosis, remains a leading infectious disease despite the availability of chemotherapy and BCG vaccine. The commonly used avirulent M. tuberculosis strain H37Ra was derived from virulent strain H37 in 1935 but the basis of virulence attenuation has remained obscure despite numerous studies. We determined the complete genomic sequence of H37Ra ATCC25177 and compared that with its virulent counterpart H37Rv and a clinical isolate CDC1551. The H37Ra genome is highly similar to that of H37Rv with respect to gene content and order but is 8,445 bp larger as a result of 53 insertions and 21 deletions in H37Ra relative to H37Rv. Variations in repetitive sequences such as IS6110 and PE/PPE/PE-PGRS family genes are responsible for most of the gross genetic changes. A total of 198 single nucleotide variations (SNVs) that are different between H37Ra and H37Rv were identified, yet 119 of them are identical between H37Ra and CDC1551 and 3 are due to H37Rv strain variation, leaving only 76 H37Ra-specific SNVs that affect only 32 genes. The biological impact of missense mutations in protein coding sequences was analyzed in silico while nucleotide variations in potential promoter regions of several important genes were verified by quantitative RT-PCR. Mutations affecting transcription factors and/or global metabolic regulations related to in vitro survival under aging stress, and mutations affecting cell envelope, primary metabolism, in vivo growth as well as variations in the PE/PPE/PE-PGRS family genes, may underlie the basis of virulence attenuation. These findings have implications not only for improved understanding of pathogenesis of M. tuberculosis but also for development of new vaccines and new therapeutic agents.
Conflict of interest statement
Figures
Similar articles
-
Differential genome organization revealed by comparative topological analysis of Mycobacterium tuberculosis strains H37Rv and H37Ra.mSystems. 2025 May 20;10(5):e0056224. doi: 10.1128/msystems.00562-24. Epub 2025 Apr 7. mSystems. 2025. PMID: 40192326 Free PMC article.
-
Genomic analysis reveals variation between Mycobacterium tuberculosis H37Rv and the attenuated M. tuberculosis H37Ra strain.Infect Immun. 1999 Nov;67(11):5768-74. doi: 10.1128/IAI.67.11.5768-5774.1999. Infect Immun. 1999. PMID: 10531227 Free PMC article.
-
SMRT genome assembly corrects reference errors, resolving the genetic basis of virulence in Mycobacterium tuberculosis.BMC Genomics. 2017 Apr 17;18(1):302. doi: 10.1186/s12864-017-3687-5. BMC Genomics. 2017. PMID: 28415976 Free PMC article.
-
Limitations of the Mycobacterium tuberculosis reference genome H37Rv in the detection of virulence-related loci.Genomics. 2017 Oct;109(5-6):471-474. doi: 10.1016/j.ygeno.2017.07.004. Epub 2017 Jul 22. Genomics. 2017. PMID: 28743540 Review.
-
An overview to understand the role of PE_PGRS family proteins in Mycobacterium tuberculosis H37 Rv and their potential as new drug targets.Biotechnol Appl Biochem. 2015 Mar-Apr;62(2):145-53. doi: 10.1002/bab.1266. Epub 2014 Nov 11. Biotechnol Appl Biochem. 2015. PMID: 24975480 Review.
Cited by
-
Application of combined CRISPR screening for genetic and chemical-genetic interaction profiling in Mycobacterium tuberculosis.Sci Adv. 2022 Nov 25;8(47):eadd5907. doi: 10.1126/sciadv.add5907. Epub 2022 Nov 23. Sci Adv. 2022. PMID: 36417506 Free PMC article.
-
Andrographolide Suppresses Pyroptosis in Mycobacterium tuberculosis-Infected Macrophages via the microRNA-155/Nrf2 Axis.Oxid Med Cell Longev. 2022 Apr 28;2022:1885066. doi: 10.1155/2022/1885066. eCollection 2022. Oxid Med Cell Longev. 2022. PMID: 35528511 Free PMC article.
-
Mitochondrial reactive oxygen species: double agents in Mycobacterium tuberculosis infection.Curr Opin Immunol. 2023 Oct;84:102366. doi: 10.1016/j.coi.2023.102366. Epub 2023 Jul 13. Curr Opin Immunol. 2023. PMID: 37453340 Free PMC article. Review.
-
Comparison of membrane proteins of Mycobacterium tuberculosis H37Rv and H37Ra strains.BMC Microbiol. 2011 Jan 24;11:18. doi: 10.1186/1471-2180-11-18. BMC Microbiol. 2011. PMID: 21261938 Free PMC article.
-
A Molecular Biological and Biochemical Investigation on Mycobacterium tuberculosis MutT Protein.Jundishapur J Microbiol. 2014 Mar;7(3):e9367. doi: 10.5812/jjm.9367. Epub 2014 Mar 1. Jundishapur J Microbiol. 2014. PMID: 25147690 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
