

Identifying functional modules in protein-protein interaction networks: an integrated exact approach
- PMID: 18586718
- PMCID: PMC2718639
- DOI: 10.1093/bioinformatics/btn161
Identifying functional modules in protein-protein interaction networks: an integrated exact approach
Abstract
Motivation: With the exponential growth of expression and protein-protein interaction (PPI) data, the frontier of research in systems biology shifts more and more to the integrated analysis of these large datasets. Of particular interest is the identification of functional modules in PPI networks, sharing common cellular function beyond the scope of classical pathways, by means of detecting differentially expressed regions in PPI networks. This requires on the one hand an adequate scoring of the nodes in the network to be identified and on the other hand the availability of an effective algorithm to find the maximally scoring network regions. Various heuristic approaches have been proposed in the literature.
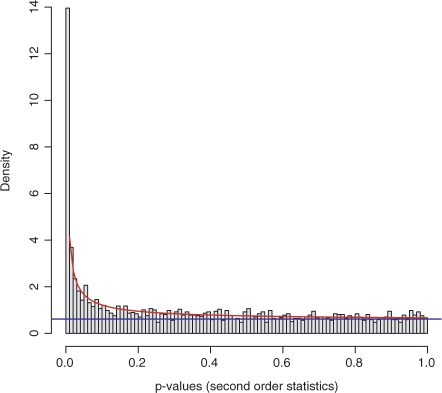
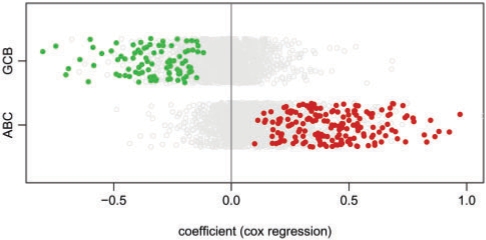
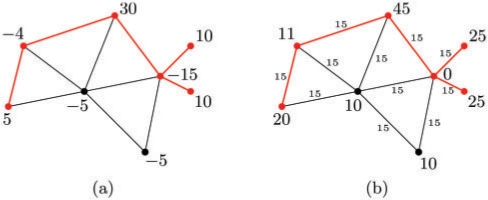
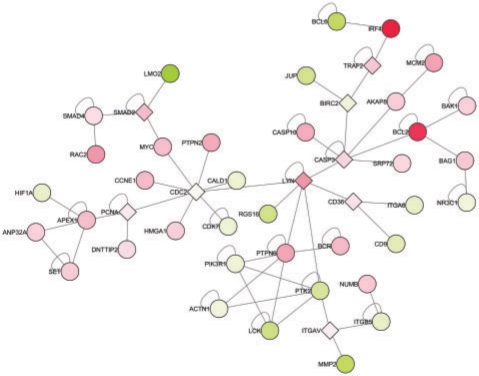
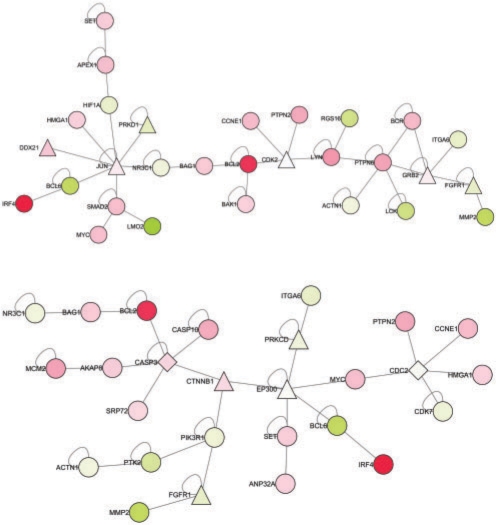
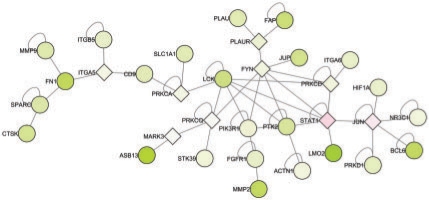
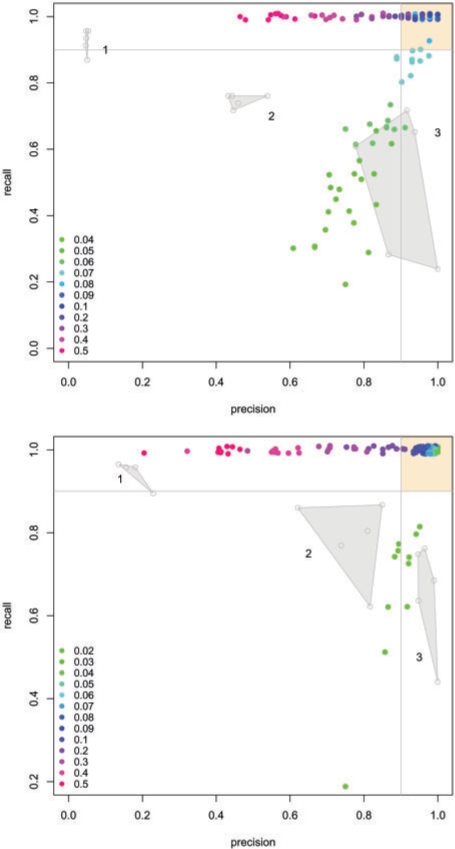
Results: Here we present the first exact solution for this problem, which is based on integer-linear programming and its connection to the well-known prize-collecting Steiner tree problem from Operations Research. Despite the NP-hardness of the underlying combinatorial problem, our method typically computes provably optimal subnetworks in large PPI networks in a few minutes. An essential ingredient of our approach is a scoring function defined on network nodes. We propose a new additive score with two desirable properties: (i) it is scalable by a statistically interpretable parameter and (ii) it allows a smooth integration of data from various sources. We apply our method to a well-established lymphoma microarray dataset in combination with associated survival data and the large interaction network of HPRD to identify functional modules by computing optimal-scoring subnetworks. In particular, we find a functional interaction module associated with proliferation over-expressed in the aggressive ABC subtype as well as modules derived from non-malignant by-stander cells.
Availability: Our software is available freely for non-commercial purposes at http://www.planet-lisa.net.
Figures
References
-
- Aittokallio T, Schwikowski B. Graph-based methods for analysing networks in cell biology. Brief. Bioinform. 2006;7:243–255. - PubMed
-
- Andersen P, Gill R. Cox's regression model for counting processes: a large sample study. Ann. Stat. 1982;10:1100–1120.
-
- Byrd RH, et al. A limited memory algorithm for bound constrained optimization. SIAM J. Sci. Comput. 1995;16:1190–1208.
-
- Cabusora L, et al. Differential network expression during drug and stress response. Bioinformatics. 2005;21:2898–2905. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
