

Prediction of drug-target interaction networks from the integration of chemical and genomic spaces
- PMID: 18586719
- PMCID: PMC2718640
- DOI: 10.1093/bioinformatics/btn162
Prediction of drug-target interaction networks from the integration of chemical and genomic spaces
Abstract
Motivation: The identification of interactions between drugs and target proteins is a key area in genomic drug discovery. Therefore, there is a strong incentive to develop new methods capable of detecting these potential drug-target interactions efficiently.
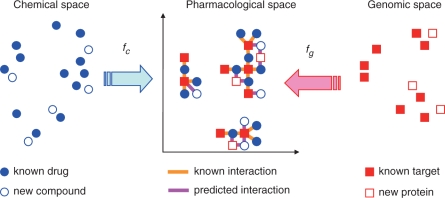
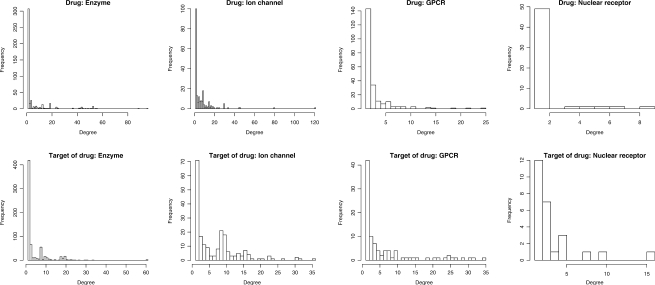
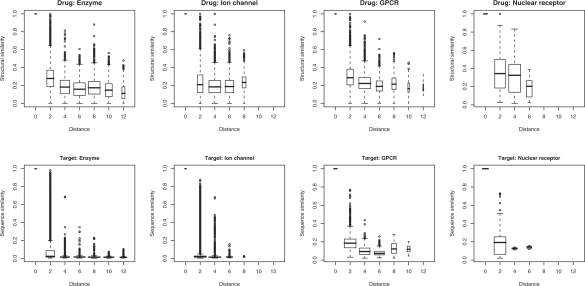
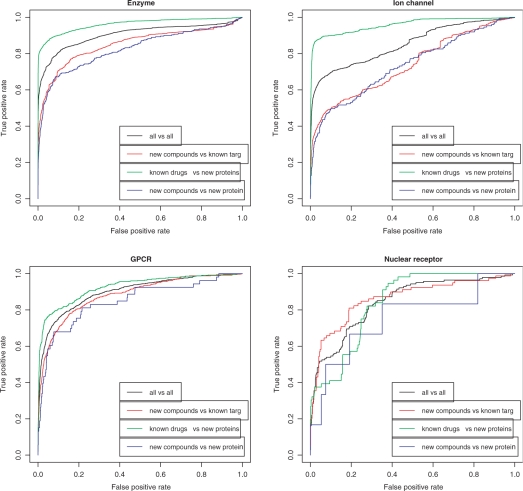
Results: In this article, we characterize four classes of drug-target interaction networks in humans involving enzymes, ion channels, G-protein-coupled receptors (GPCRs) and nuclear receptors, and reveal significant correlations between drug structure similarity, target sequence similarity and the drug-target interaction network topology. We then develop new statistical methods to predict unknown drug-target interaction networks from chemical structure and genomic sequence information simultaneously on a large scale. The originality of the proposed method lies in the formalization of the drug-target interaction inference as a supervised learning problem for a bipartite graph, the lack of need for 3D structure information of the target proteins, and in the integration of chemical and genomic spaces into a unified space that we call 'pharmacological space'. In the results, we demonstrate the usefulness of our proposed method for the prediction of the four classes of drug-target interaction networks. Our comprehensively predicted drug-target interaction networks enable us to suggest many potential drug-target interactions and to increase research productivity toward genomic drug discovery.
Availability: Softwares are available upon request.
Supplementary information: Datasets and all prediction results are available at http://web.kuicr.kyoto-u.ac.jp/supp/yoshi/drugtarget/.
Figures
Similar articles
-
Drug-target interaction prediction from chemical, genomic and pharmacological data in an integrated framework.Bioinformatics. 2010 Jun 15;26(12):i246-54. doi: 10.1093/bioinformatics/btq176. Bioinformatics. 2010. PMID: 20529913 Free PMC article.
-
Supervised prediction of drug-target interactions using bipartite local models.Bioinformatics. 2009 Sep 15;25(18):2397-403. doi: 10.1093/bioinformatics/btp433. Epub 2009 Jul 15. Bioinformatics. 2009. PMID: 19605421 Free PMC article.
-
[Prediction of network drug target based on improved model of bipartite graph valuation].Zhongguo Zhong Yao Za Zhi. 2012 Jan;37(2):125-9. Zhongguo Zhong Yao Za Zhi. 2012. PMID: 22737836 Chinese.
-
The Computational Models of Drug-target Interaction Prediction.Protein Pept Lett. 2020;27(5):348-358. doi: 10.2174/0929866526666190410124110. Protein Pept Lett. 2020. PMID: 30968771 Review.
-
Molecular recognition and binding free energy calculations in drug development.Curr Pharm Biotechnol. 2008 Apr;9(2):87-95. doi: 10.2174/138920108783955155. Curr Pharm Biotechnol. 2008. PMID: 18393865 Review.
Cited by
-
AutoDTI++: deep unsupervised learning for DTI prediction by autoencoders.BMC Bioinformatics. 2021 Apr 20;22(1):204. doi: 10.1186/s12859-021-04127-2. BMC Bioinformatics. 2021. PMID: 33879050 Free PMC article.
-
Effects of protein interaction data integration, representation and reliability on the use of network properties for drug target prediction.BMC Bioinformatics. 2012 Nov 12;13:294. doi: 10.1186/1471-2105-13-294. BMC Bioinformatics. 2012. PMID: 23146171 Free PMC article.
-
Prediction of compound-target interactions of natural products using large-scale drug and protein information.BMC Bioinformatics. 2016 Jul 28;17 Suppl 6(Suppl 6):219. doi: 10.1186/s12859-016-1081-y. BMC Bioinformatics. 2016. PMID: 27490208 Free PMC article.
-
Exploring the ligand-protein networks in traditional chinese medicine: current databases, methods, and applications.Evid Based Complement Alternat Med. 2013;2013:806072. doi: 10.1155/2013/806072. Epub 2013 Jun 2. Evid Based Complement Alternat Med. 2013. PMID: 23818932 Free PMC article.
-
Netpredictor: R and Shiny package to perform drug-target network analysis and prediction of missing links.BMC Bioinformatics. 2018 Jul 16;19(1):265. doi: 10.1186/s12859-018-2254-7. BMC Bioinformatics. 2018. PMID: 30012095 Free PMC article.
References
-
- Cheng AC, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007;25:71–75. - PubMed
-
- Dobson CM. Chemical space and biology. Nature. 2004;432:824–828. - PubMed
-
- Gribskov M, Robinson NL. Use of receiver operating characteristic (roc) analysis to evaluate sequence matching. Comput. Chem. 1996;20:25–33. - PubMed
-
- Haggarty SJ, et al. Multidimensional chemical genetic analysis of diversity-oriented synthesis-derived deacetylase inhibitors using cell-based assays. Chem. Biol. 2003;10:383–396. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
