

Review
doi: 10.1161/CIRCULATIONAHA.108.776831.
Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics
Affiliations
- PMID: 18591451
- PMCID: PMC2735334
- DOI: 10.1161/CIRCULATIONAHA.108.776831
Item in Clipboard
Review
Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics
Circulation.
.
No abstract available
Conflict of interest statement
None
Figures
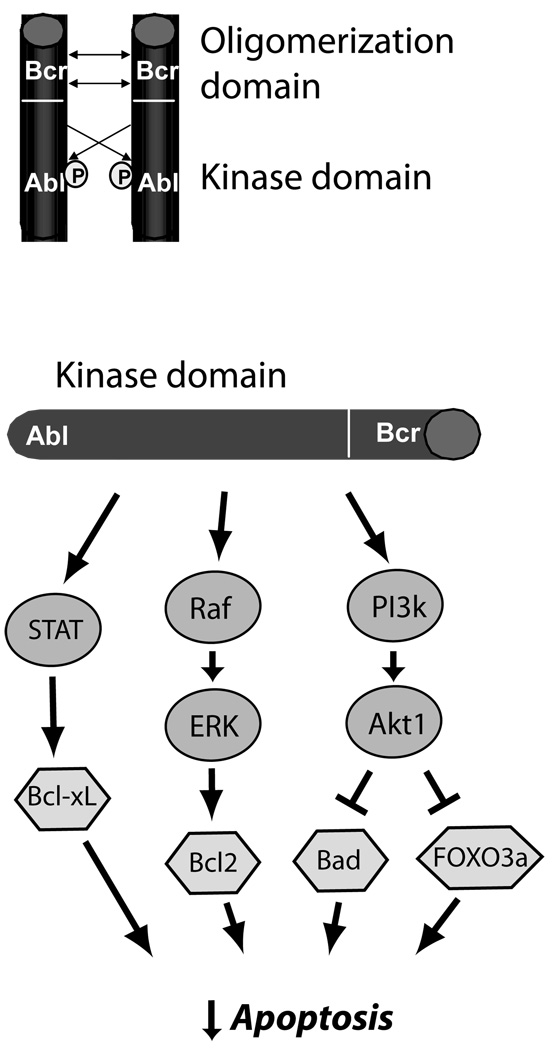
Mechanisms of carcinogenesis of Bcr-Abl in CML. Oligomerization and cross-phosphorylation (P) of Bcr-Abl fusion proteins (top) leads to constitutive activation of the Abl kinase domain. This leads (bottom) to activation of three key pro-survival pathways, STAT5 (signal transducer and activator of transcription 5), which leads to increased expression of anti-apoptotic Bcl-xL, the Raf → ERK (extracellular signal-regulated kinase, or MAP kinase) pathway, which increases expression of anti-apoptotic Bcl2, and the phosphoinositide-3 kinase → Akt pathway, a major anti-apoptotic pathway in cancer cells and cardiomyocytes, that inhibits pro-apoptotic factors FOXO3a and Bad. This culminates in potent inhibition of apoptosis in CML cells.
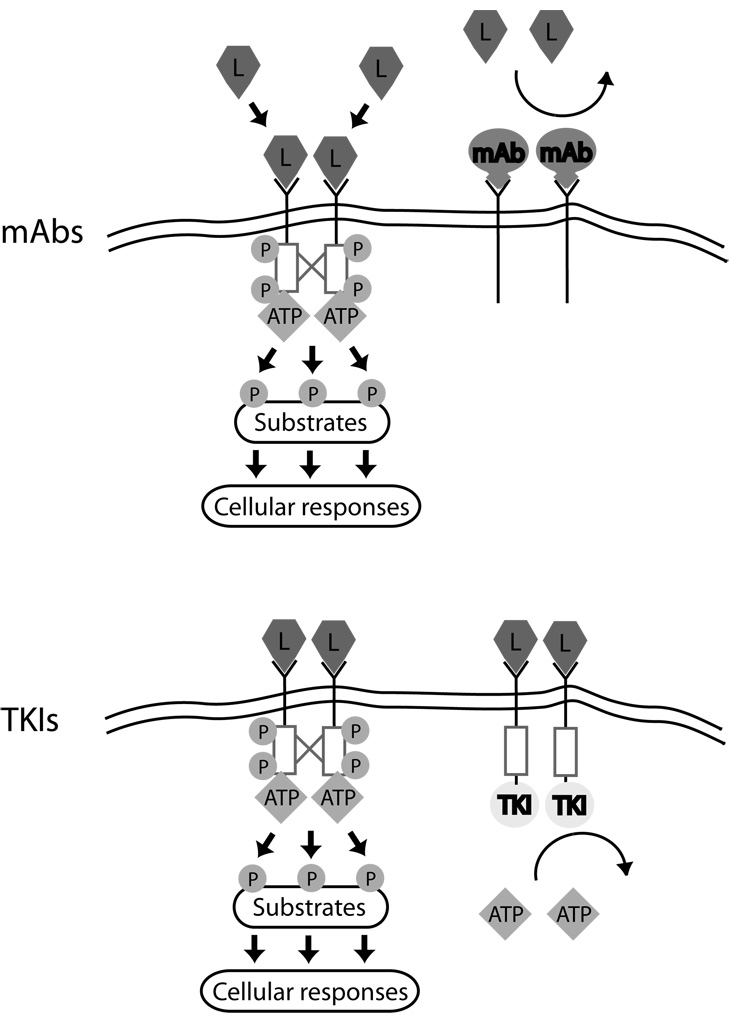
Mechanisms of action of monoclonal antibodies vs. small molecule tyrosine kinase inhibitors (TKIs). Ligand (L) binding to RTKs leads to receptor dimerization, cross phosphorylation (red lines and P), and activation of the intracellular tyrosine kinase domain (red boxes). Substrates are then phosphorylated, leading to cellular responses. Monoclonal antibodies (mAbs, Top) interfere with ligand binding to receptor and/or receptor dimerization and cross-phosphorylation, blocking activation of the RTKs. TKIs (Bottom) do not prevent ligand binding or dimerization, but by preventing ATP from binding to the kinase domain, they block cross-phosphorylation of receptors and phosphorylation of substrates.
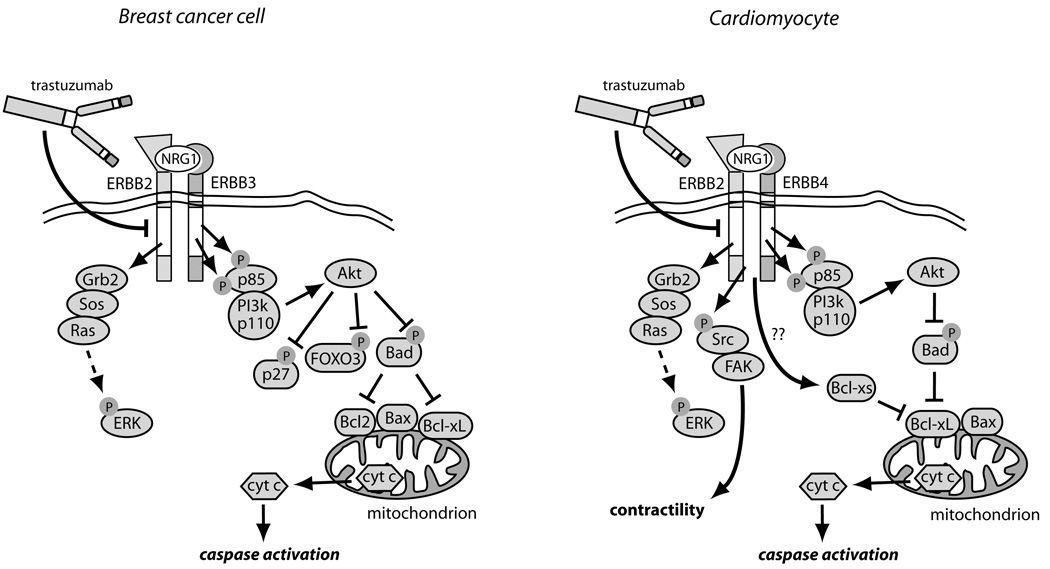
Comparison of ERBB2 signaling and its inhibition by trastuzumab in breast cancer cells vs. cardiomyocytes. In breast cancer cells over-expressing ERBB2, ERBB2 homodimers or ERBB2/ERBB3 heterodimers form, leading to constitutive activation of the ERK, PI3K/Akt, and STAT3 pathways (latter not shown). Akt blocks apoptosis by phosphorylating and inhibiting two key pro-apoptotic factors, Bad and FOXO3A, and also inactivates the cyclin-dependent kinase inhibitor, p27, thereby enhancing cell proliferation. Trastuzumab blocks all downstream signaling, but particularly important may be reversing the inhibition of Bad, leading to activation of Bax, cyctochrome c release and apoptosis. In cardiomyocytes exposed to Nrg1, ERBB2/ERBB4 heterodimers form, again activating ERK and Akt. Trastuzumab blocks this activation and, via multiple mechanisms including alterations in levels of Bcl-X family members, leads to mitochondrial dysfunction, energy compromise, and cytochrome c release. Trastuzumab also blocks Nrg1-mediated activation of Src and Fak, and this appears to worsen LV dysfunction.
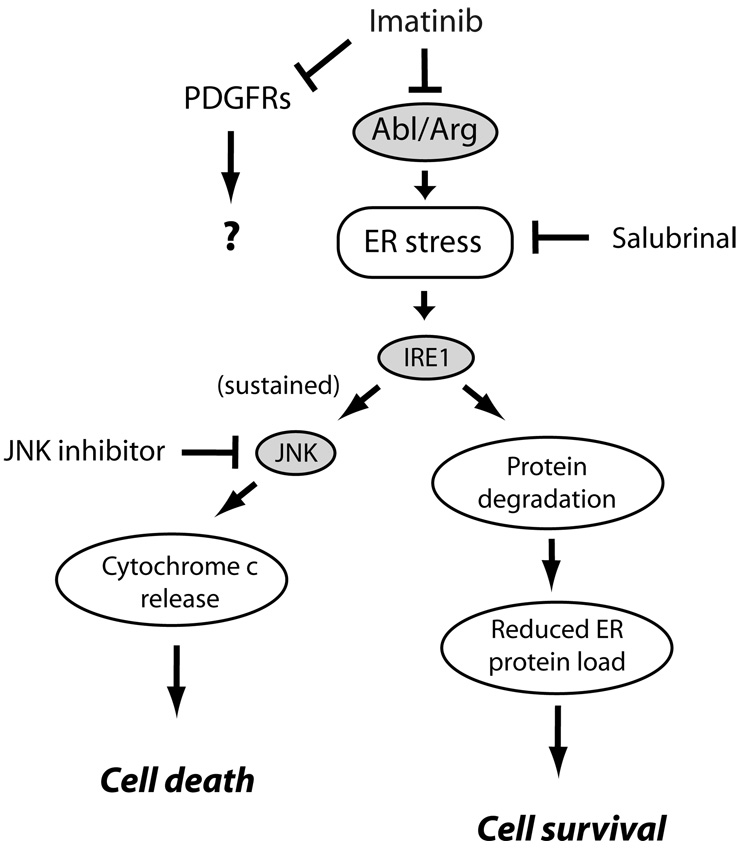
Pathways of imatinib-induced cardiomyocyte toxicity. Imatinib, via inhibition of Abl/Arg, leads via unclear mechanisms to induction of ER stress. This activates protein kinase IRE1 which upregulates factors involved in degradation of mis-folded proteins in the ER, thereby attempting to restore homeostasis. If ER stress is sustained, however, JNKs are activated leading to activation of the intrinsic apoptosis program and cell death. Both salubrinal, an inhibitor of ER stress, and JNK inhibition by a peptide antagonist, protected from imatinib cardiomyocyte toxicity. The role, if any, of inhibition of PDGFRs in imatinib-induced cardiotoxicity is unknown at this time.
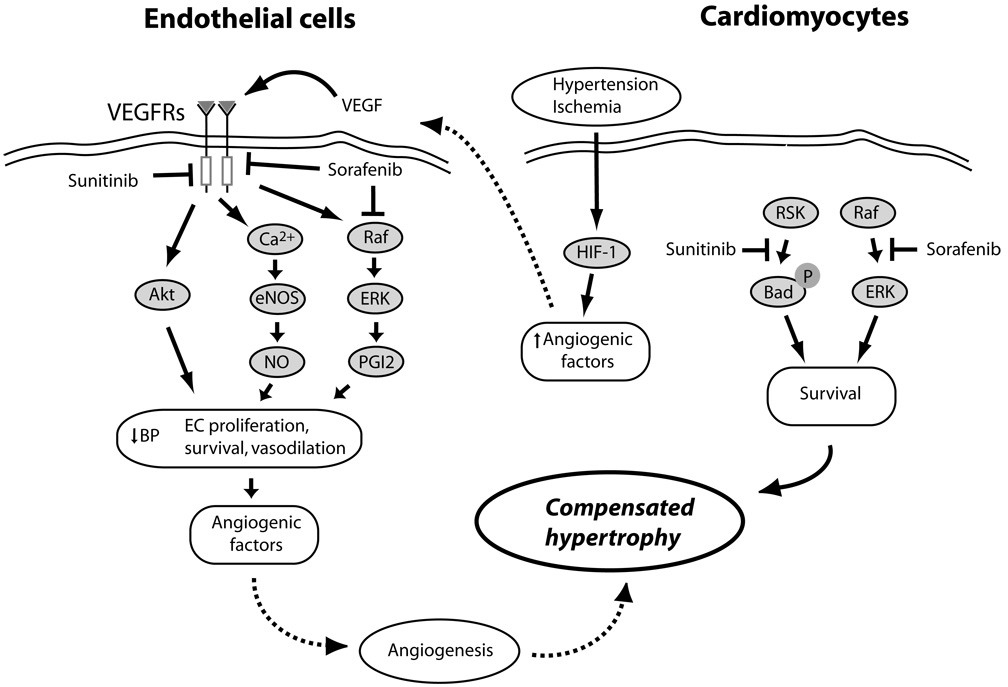
Mechanisms of VEGF/VEGFR-mediated protection of cardiomyocytes from pressure stress and ischemia, and potential interactions of sunitinib and sorafenib. Hypertensive stress in the heart and the resulting relative ischemia/hypoxia of cardiomyocytes activate HIF-1, leading to the release of angiogenic factors including VEGF. VEGF activates VEGFRs on the surface of endothelial cells (EC), activating a number of pathways that induce vasodilation (NO and PGI2), EC proliferation and survival (Akt and ERKs), and the further release of proangiogenic factors, all of which lead to angiogenesis in the heart, allowing compensated hypertrophy to occur., Sunitinib and sorafenib inhibit VEGFRs in ECs, potentially blocking angiogenesis and leading to decompensated hypertrophy. Sunitinib, via an off-target effect, may also inhibit various cytoprotective pathways in the heart (e.g. RSK, which would otherwise inhibit pro-apoptotic Bad). Sorafenib, by inhibiting the Raf/ERK pathway, could also induce apoptosis in ECs and cardiomyocytes.
References
-
- Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–1338. - PubMed
-
- Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, Figlin RA. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007;356:115–124. - PubMed
-
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004;350:2335–2342. - PubMed
-
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001;344:783–792. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
