

Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex
- PMID: 18591664
- PMCID: PMC2442127
- DOI: 10.1073/pnas.0801294105
Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex
Abstract
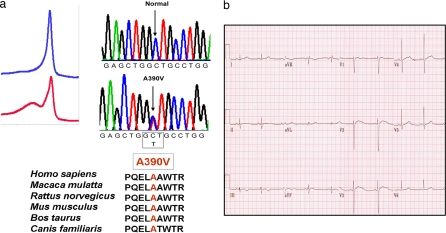
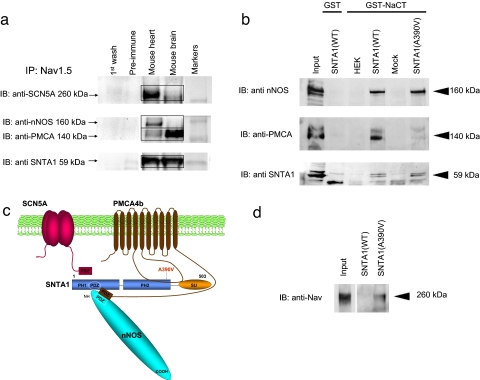
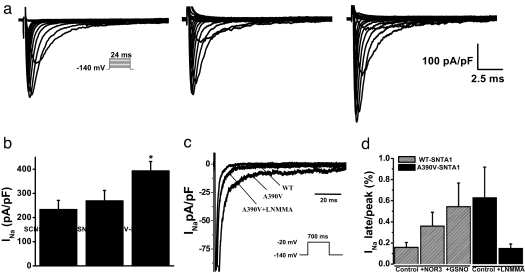
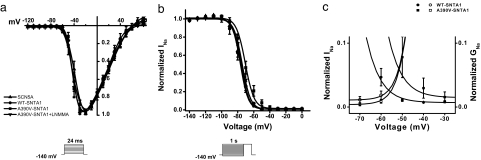
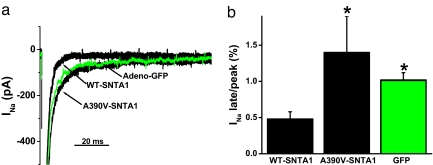
Mutations in 11 genes that encode ion channels or their associated proteins cause inherited long QT syndrome (LQTS) and account for approximately 75-80% of cases (LQT1-11). Direct sequencing of SNTA1, the gene encoding alpha1-syntrophin, was performed in a cohort of LQTS patients that were negative for mutations in the 11 known LQTS-susceptibility genes. A missense mutation (A390V-SNTA1) was found in a patient with recurrent syncope and markedly prolonged QT interval (QTc, 530 ms). SNTA1 links neuronal nitric oxide synthase (nNOS) to the nNOS inhibitor plasma membrane Ca-ATPase subtype 4b (PMCA4b); SNTA1 also is known to associate with the cardiac sodium channel SCN5A. By using a GST-fusion protein of the C terminus of SCN5A, we showed that WT-SNTA1 interacted with SCN5A, nNOS, and PMCA4b. In contrast, A390V-SNTA1 selectively disrupted association of PMCA4b with this complex and increased direct nitrosylation of SCN5A. A390V-SNTA1 expressed with SCN5A, nNOS, and PMCA4b in heterologous cells increased peak and late sodium current compared with WT-SNTA1, and the increase was partially inhibited by NOS blockers. Expression of A390V-SNTA1 in cardiac myocytes also increased late sodium current. We conclude that the A390V mutation disrupted binding with PMCA4b, released inhibition of nNOS, caused S-nitrosylation of SCN5A, and was associated with increased late sodium current, which is the characteristic biophysical dysfunction for sodium-channel-mediated LQTS (LQT3). These results establish an SNTA1-based nNOS complex attached to SCN5A as a key regulator of sodium current and suggest that SNTA1 be considered a rare LQTS-susceptibility gene.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Lehnart SE, et al. Inherited arrhythmias: A National Heart, Lung, and Blood Institute and Office of Rare Diseases workshop consensus report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene mutations affecting ion channel function. Circulation. 2007;116:2325–2345. - PubMed
-
- Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc Med. 2005;15:35–40. - PubMed
-
- Vatta M, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
