

EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer
- PMID: 18594010
- PMCID: PMC3025451
- DOI: 10.1158/1078-0432.CCR-08-0168
EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer
Abstract
Purpose: The EML4-ALK fusion gene has been detected in approximately 7% of Japanese non-small cell lung cancers (NSCLC). We determined the frequency of EML4-ALK in Caucasian NSCLC and in NSCLC cell lines. We also determined whether TAE684, a specific ALK kinase inhibitor, would inhibit the growth of EML4-ALK-containing cell lines in vitro and in vivo.
Experimental design: We screened 305 primary NSCLC [both U.S. (n = 138) and Korean (n = 167) patients] and 83 NSCLC cell lines using reverse transcription-PCR and by exon array analyses. We evaluated the efficacy of TAE684 against NSCLC cell lines in vitro and in vivo.
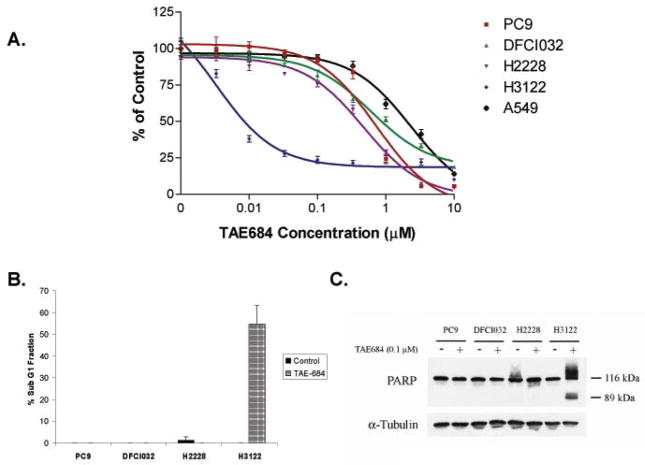
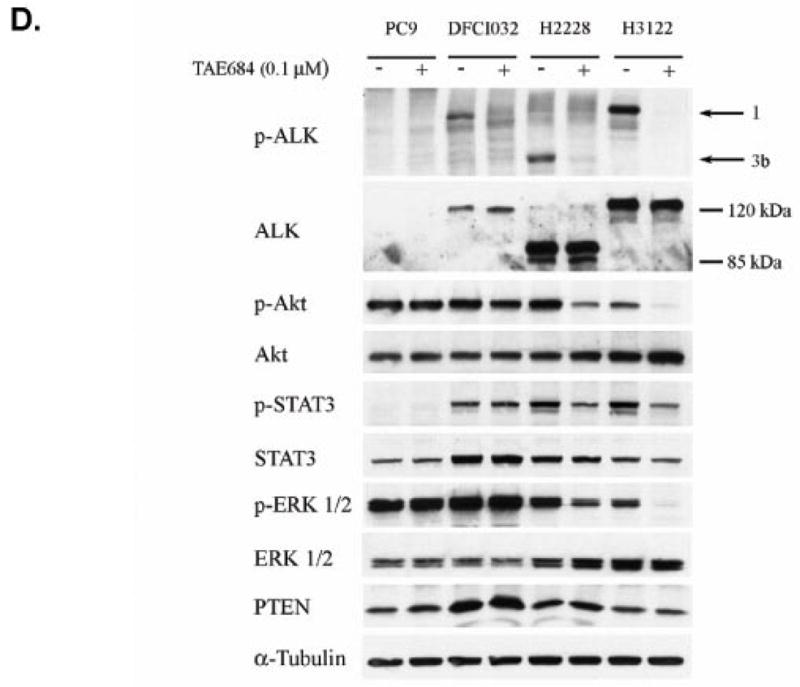
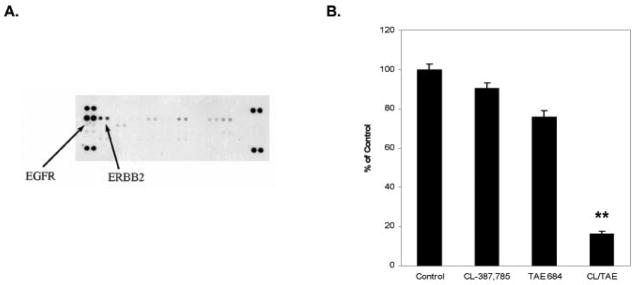
Results: We detected four different variants, including two novel variants, of EML4-ALK using reverse transcription-PCR in 8 of 305 tumors (3%) and 3 of 83 (3.6%) NSCLC cell lines. All EML4-ALK-containing tumors and cell lines were adenocarcinomas. EML4-ALK was detected more frequently in NSCLC patients who were never or light (<10 pack-years) cigarette smokers compared with current/former smokers (6% versus 1%; P = 0.049). TAE684 inhibited the growth of one of three (H3122) EML4-ALK-containing cell lines in vitro and in vivo, inhibited Akt phosphorylation, and caused apoptosis. In another EML4-ALK cell line, DFCI032, TAE684 was ineffective due to coactivation of epidermal growth factor receptor and ERBB2. The combination of TAE684 and CL-387,785 (epidermal growth factor receptor/ERBB2 kinase inhibitor) inhibited growth and Akt phosphorylation and led to apoptosis in the DFCI032 cell line.
Conclusions: EML4-ALK is found in the minority of NSCLC. ALK kinase inhibitors alone or in combination may nevertheless be clinically effective treatments for NSCLC patients whose tumors contain EML4-ALK.
Figures




References
-
- Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263:1281–4. - PubMed
-
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. - PubMed
-
- Li R, Morris SW. Development of anaplastic lymphoma kinase (ALK) small-molecule inhibitors for cancer therapy. Med Res Rev. 2007 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
