

Complement and the atypical hemolytic uremic syndrome in children
- PMID: 18594873
- PMCID: PMC6904381
- DOI: 10.1007/s00467-008-0872-4
Complement and the atypical hemolytic uremic syndrome in children
Abstract
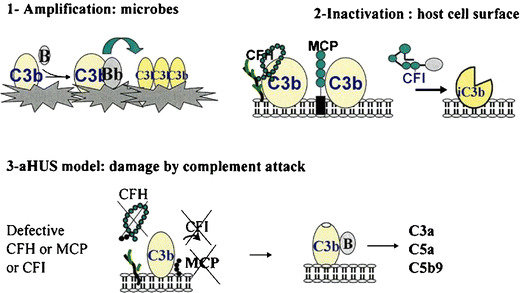
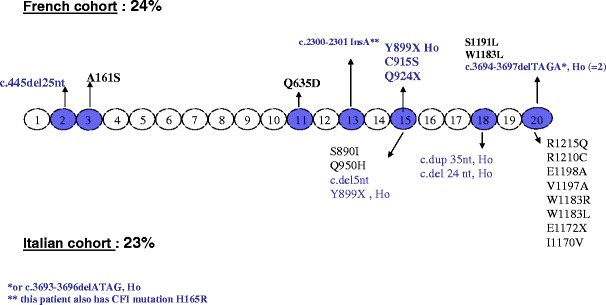
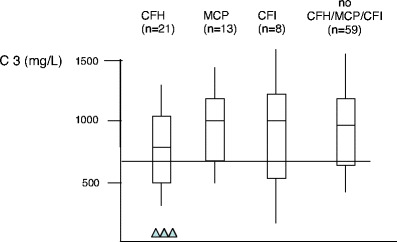
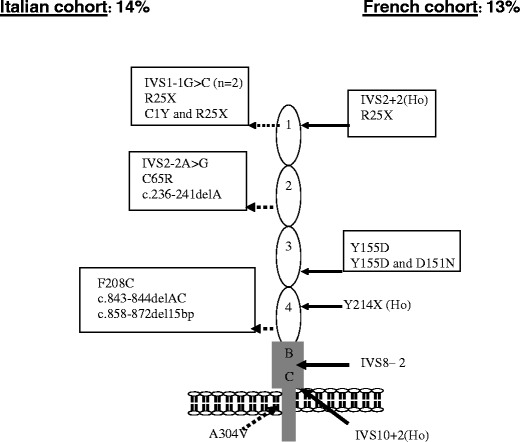
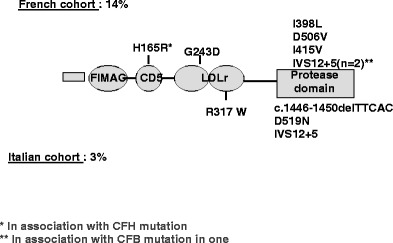
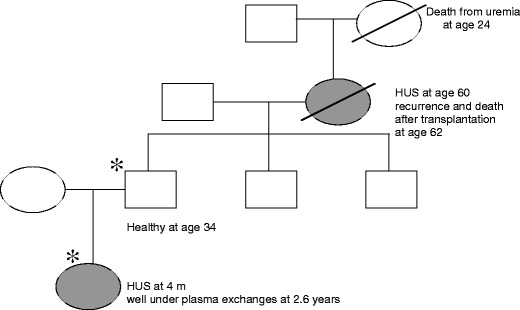
Over the past decade, atypical hemolytic uremic syndrome (aHUS) has been demonstrated to be a disorder of the regulation of the complement alternative pathway. Among approximately 200 children with the disease, reported in the literature, 50% had mutations of the complement regulatory proteins factor H, membrane cofactor protein (MCP) or factor I. Mutations in factor B and C3 have also been reported recently. In addition, 10% of children have factor H dysfunction due to anti-factor H antibodies. Early age at onset appears as characteristic of factor H and factor I mutated patients, while MCP-associated HUS is not observed before age 1 year. Low C3 level may occur in patients with factor H and factor I mutation, while C3 level is generally normal in MCP-mutated patients. Normal plasma factor H and factor I levels do not preclude the presence of a mutation in these genes. The worst prognosis is for factor H-mutated patients, as 60% die or reach end-stage renal disease (ESRD) within the first year after onset of the disease. Patients with mutations in MCP have a relapsing course, but no patient has ever reached ESRD in the first year of the disease. Half of the patients with factor I mutations have a rapid evolution to ESRD, but half recover. Early intensive plasmatherapy appears to have a beneficial effect, except in MCP-mutated patients. There is a high risk of graft loss for HUS recurrence or thrombosis in all groups except the MCP-mutated group. Recent success of liver-kidney transplantation combined with plasmatherapy opens this option for patients with mutations of factors synthesized in the liver. New therapies such as factor H concentrate or complement inhibitors offer hope for the future.
Figures
References
-
- Besbas N, Karpman D, Landau D, Loirat C, Proesmans W, Remuzzi G, Rizzoni G, Taylor CM, Van de Kar N, Zimmerhackl LB. A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int. 2006;70:423–431. - PubMed
-
- Garg AX, Suri RS, Barrowman N, Rehman F, Matsell D, Rosas-Arellano MP, Salvadori M, Haynes RB, Clark WF. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: a systematic review, meta-analysis, and meta-regression. JAMA. 2003;290:1360–1370. - PubMed
-
- Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:1035–1050. - PubMed
-
- Caprioli J, Bettinaglio P, Zipfel PF, Amadei B, Daina E, Gamba S, Skerka C, Marziliano N, Remuzzi G, Noris M. The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol. 2001;12:297–307. - PubMed
-
- Caprioli J, Castelletti F, Bucchioni S, Bettinaglio P, Bresin E, Pianetti G, Gamba S, Brioschi S, Daina E, Remuzzi G, Noris M. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. 2003;12:3385–3395. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
