

Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease
- PMID: 18596167
- PMCID: PMC2676733
- DOI: 10.1523/JNEUROSCI.0800-08.2008
Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease
Abstract
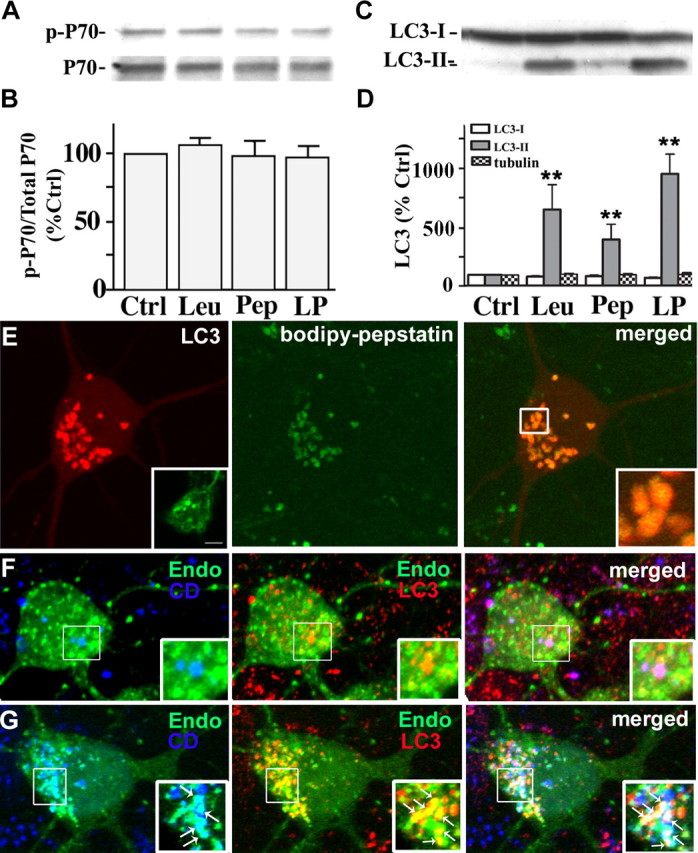
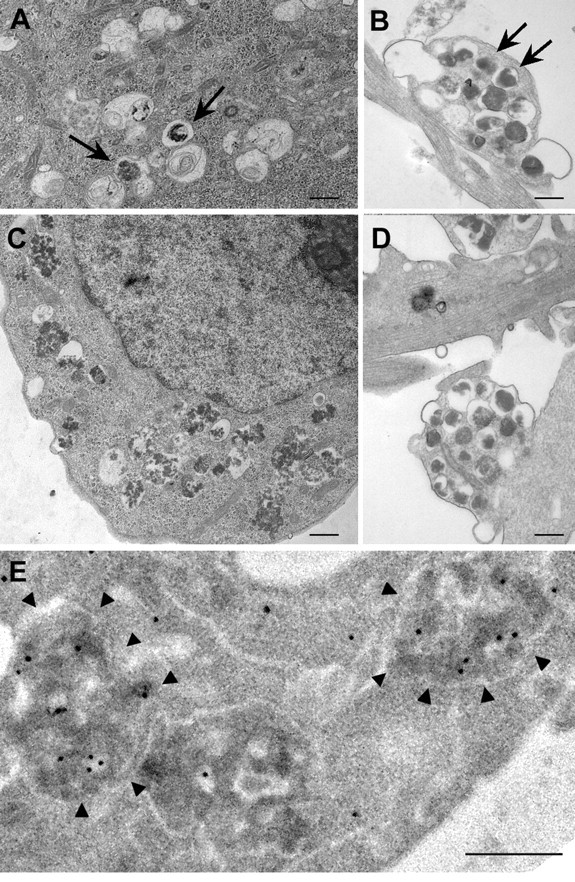
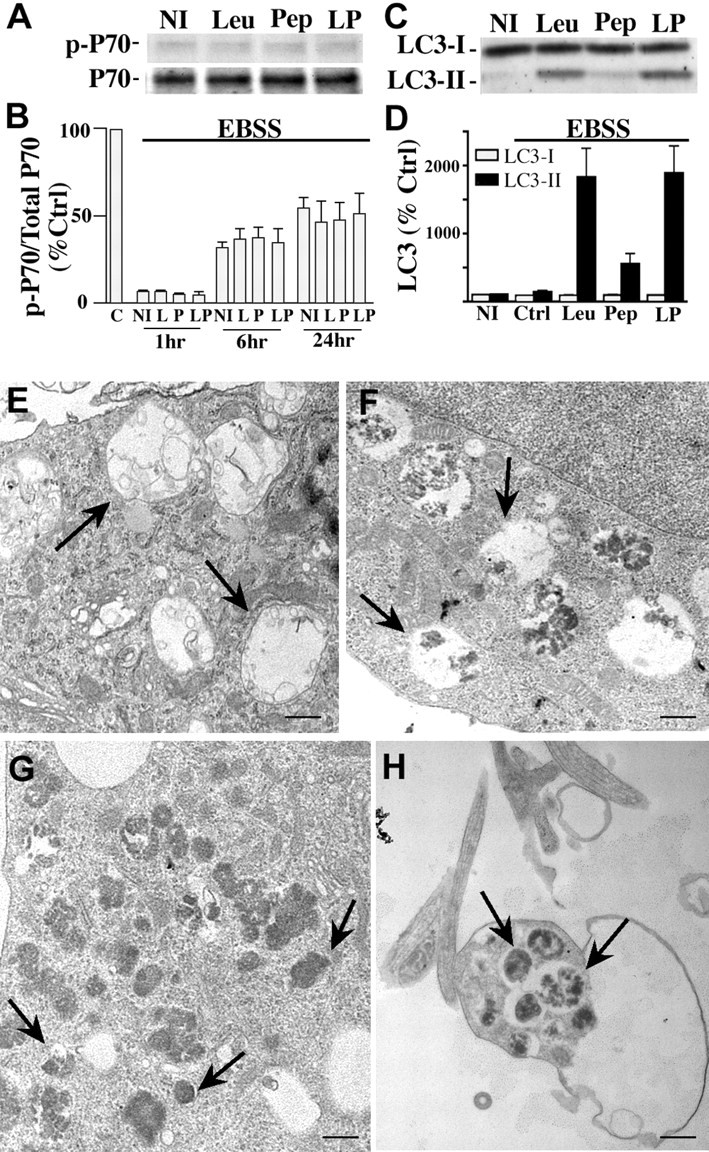
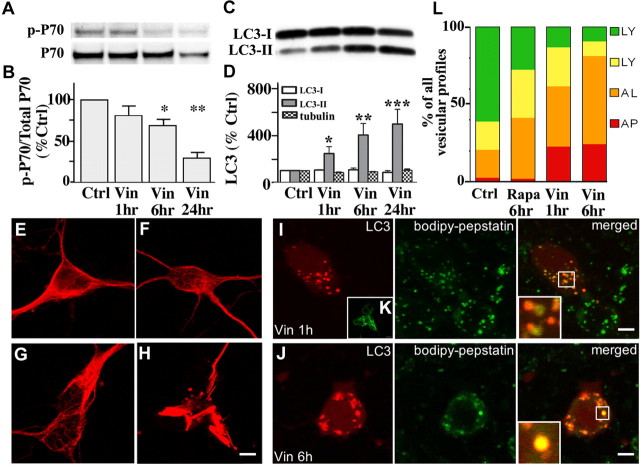
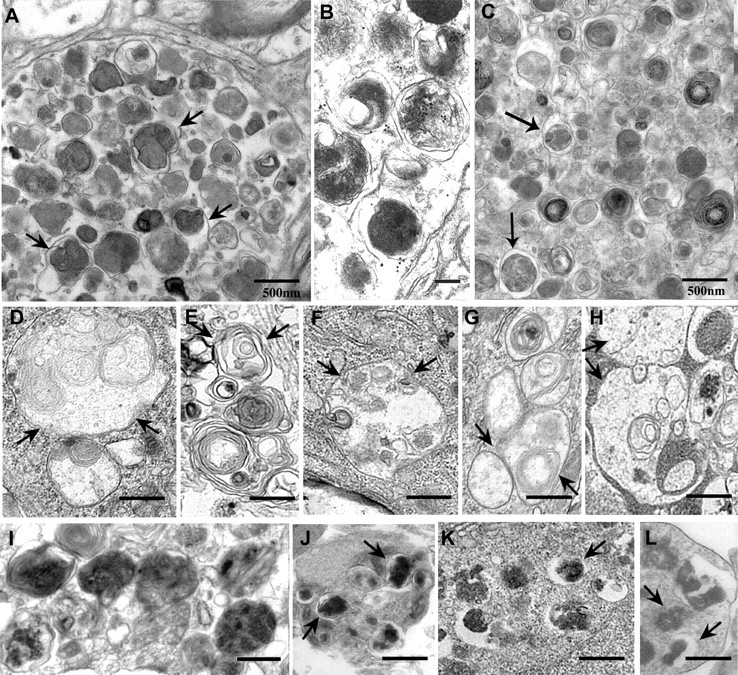
Macroautophagy, a major pathway for organelle and protein turnover, has been implicated in the neurodegeneration of Alzheimer's disease (AD). The basis for the profuse accumulation of autophagic vacuoles (AVs) in affected neurons of the AD brain, however, is unknown. In this study, we show that constitutive macroautophagy in primary cortical neurons is highly efficient, because newly formed autophagosomes are rapidly cleared by fusion with lysosomes, accounting for their scarcity in the healthy brain. Even after macroautophagy is strongly induced by suppressing mTOR (mammalian target of rapamycin) kinase activity with rapamycin or nutrient deprivation, active cathepsin-positive autolysosomes rather than LC3-II-positive autophagosomes predominate, implying efficient autophagosome clearance in healthy neurons. In contrast, selectively impeding late steps in macroautophagy by inhibiting cathepsin-mediated proteolysis within autolysosomes with cysteine- and aspartyl-protease inhibitors caused a marked accumulation of electron-dense double-membrane-limited AVs, containing cathepsin D and incompletely degraded LC3-II in perikarya and neurites. Similar structures accumulated in large numbers when fusion of autophagosomes with lysosomes was slowed by disrupting their transport on microtubules with vinblastine. Finally, we find that the autophagic vacuoles accumulating after protease inhibition or prolonged vinblastine treatment strongly resembled AVs that collect in dystrophic neurites in the AD brain and in an AD mouse model. We conclude that macroautophagy is constitutively active and highly efficient in healthy neurons and that the autophagic pathology observed in AD most likely arises from impaired clearance of AVs rather than strong autophagy induction alone. Therapeutic modulation of autophagy in AD may, therefore, require targeting late steps in the autophagic pathway.
Figures



References
-
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. - PubMed
-
- Barber AJ, Nakamura M, Wolpert EB, Reiter CE, Seigel GM, Antonetti DA, Gardner TW. Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J Biol Chem. 2001;276:32814–32821. - PubMed
-
- Berg TO, Fengsrud M, Strømhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem. 1998;273:21883–21892. - PubMed
-
- Bi X, Zhou J, Lynch G. Lysosomal protease inhibitors induce meganeurites and tangle-like structures in entorhinohippocampal regions vulnerable to Alzheimer's disease. Exp Neurol. 1999;158:312–327. - PubMed
-
- Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem. 1995;270:2320–2326. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous