

Mechanisms regulating cytochrome c release in pancreatic mitochondria
- PMID: 18596195
- PMCID: PMC4551464
- DOI: 10.1136/gut.2007.147207
Mechanisms regulating cytochrome c release in pancreatic mitochondria
Abstract
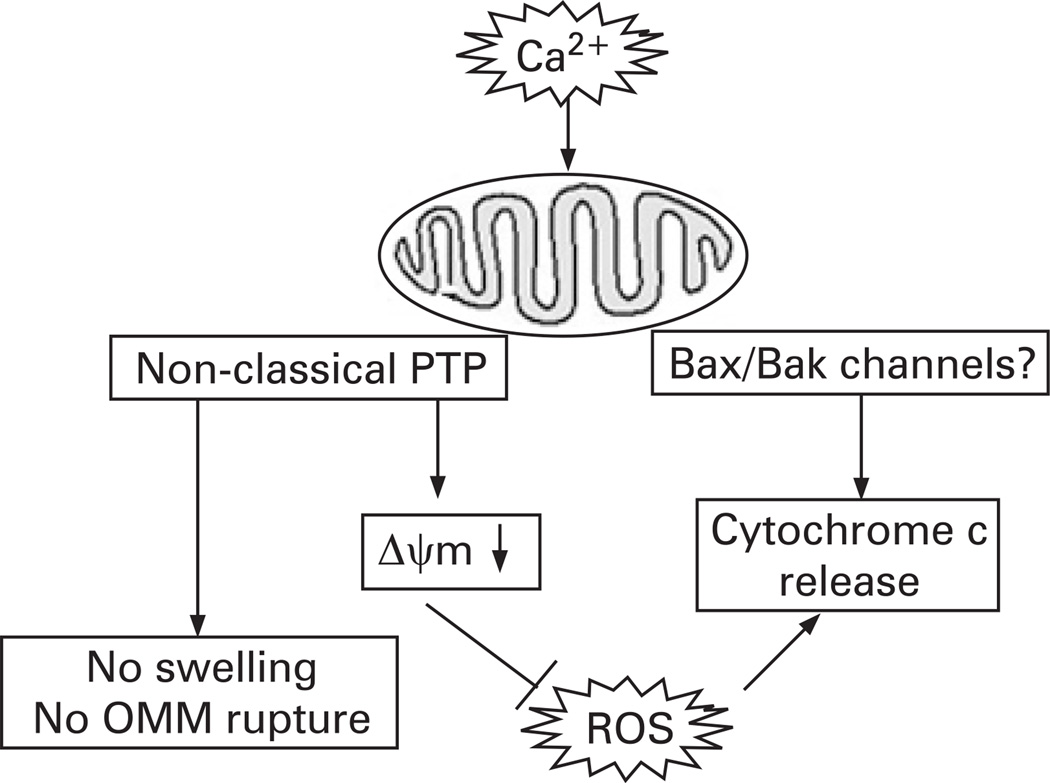
Background: Mechanisms of acinar cell death in pancreatitis are poorly understood. Cytochrome c release is a central event in apoptosis in pancreatitis. Here, we assessed the regulation of pancreatic cytochrome c release by Ca(2+), mitochondrial membrane potential (Delta Psi m), and reactive oxygen species (ROS), the signals involved in acute pancreatitis. We used both isolated rat pancreatic mitochondria and intact acinar cells hyperstimulated with cholecystokinin-8 (CCK-8; in vitro model of acute pancreatitis).
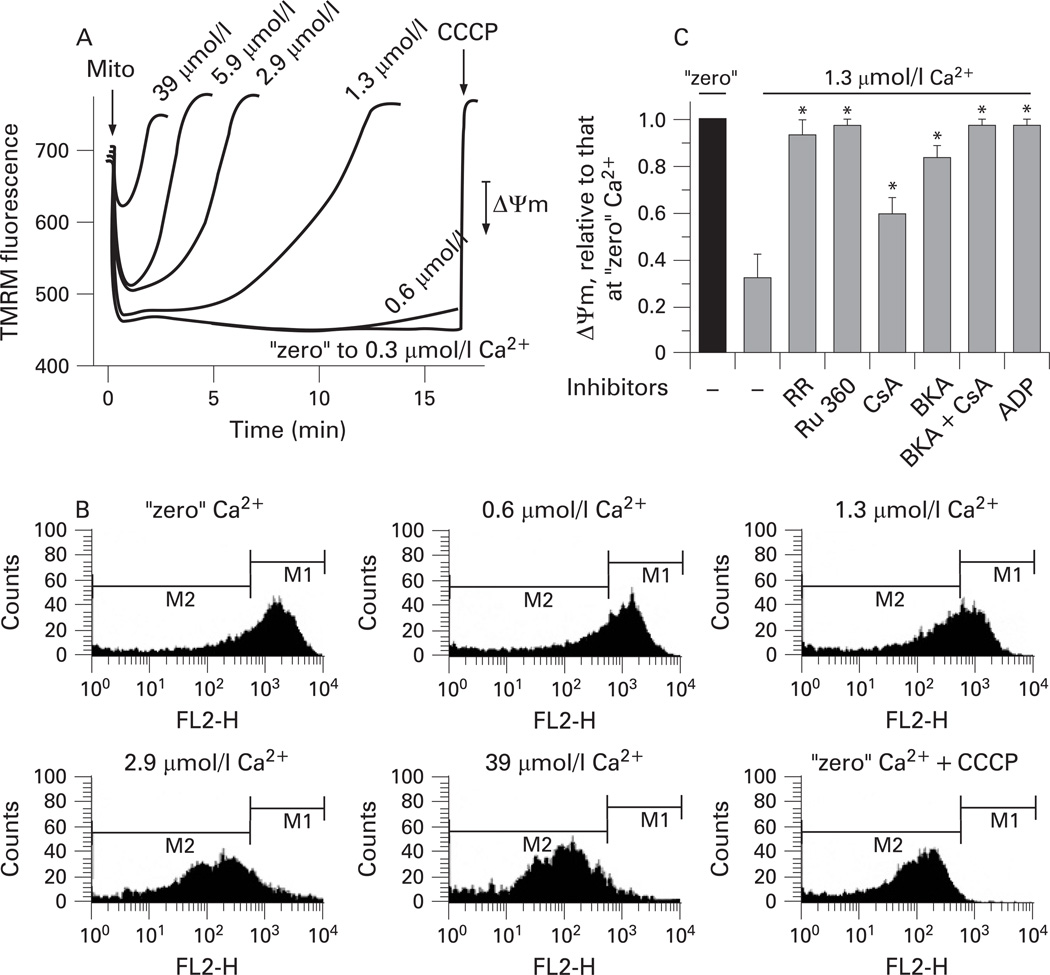
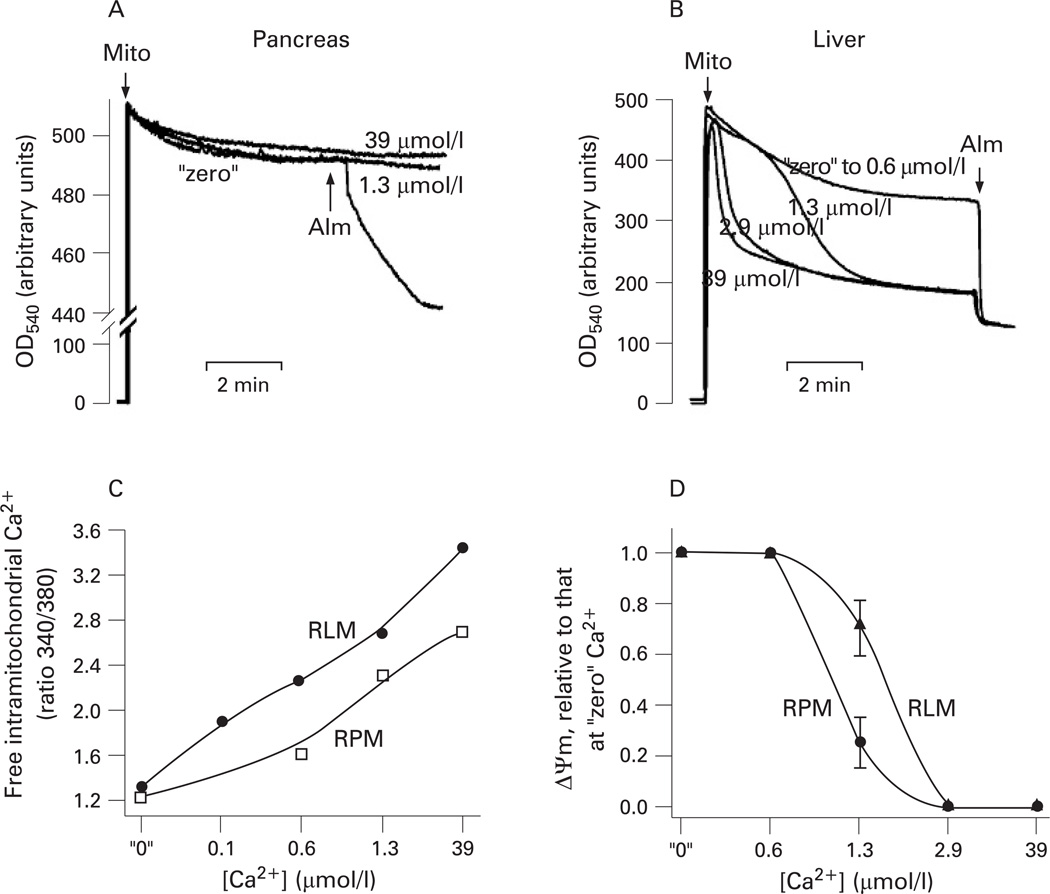
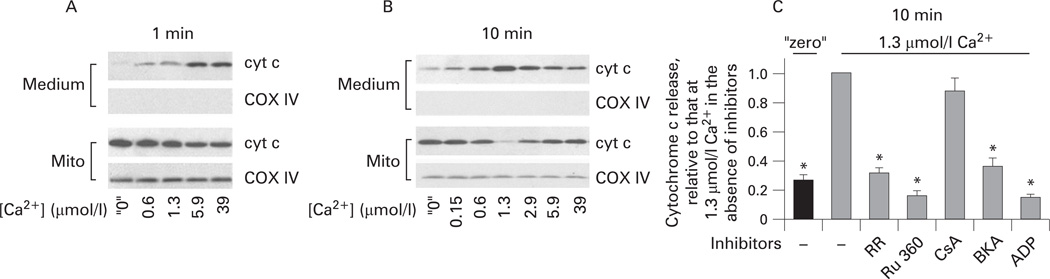
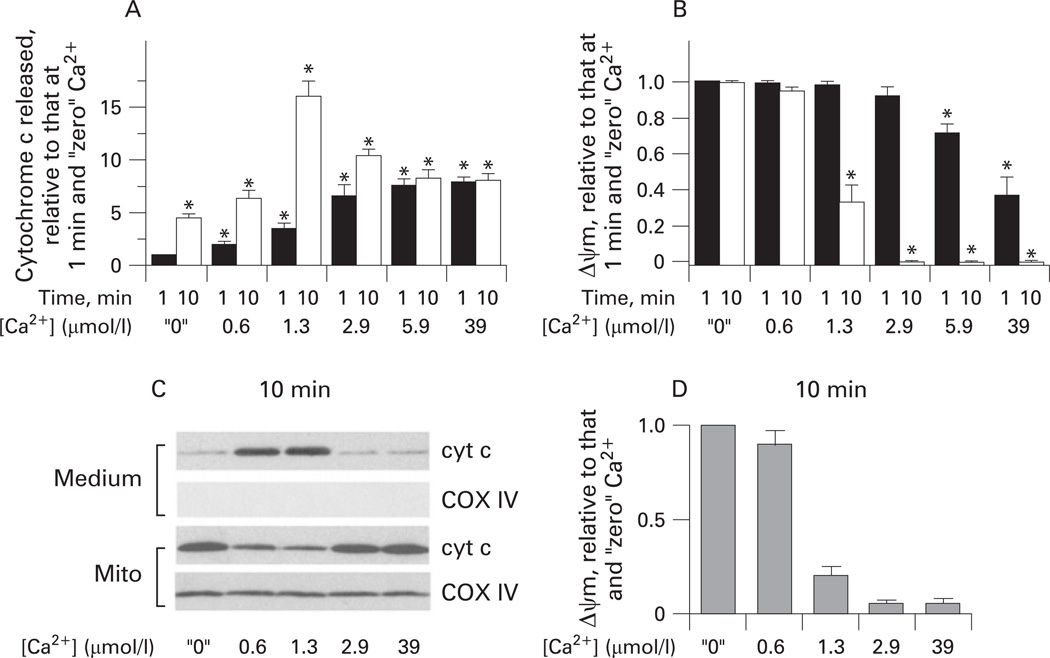
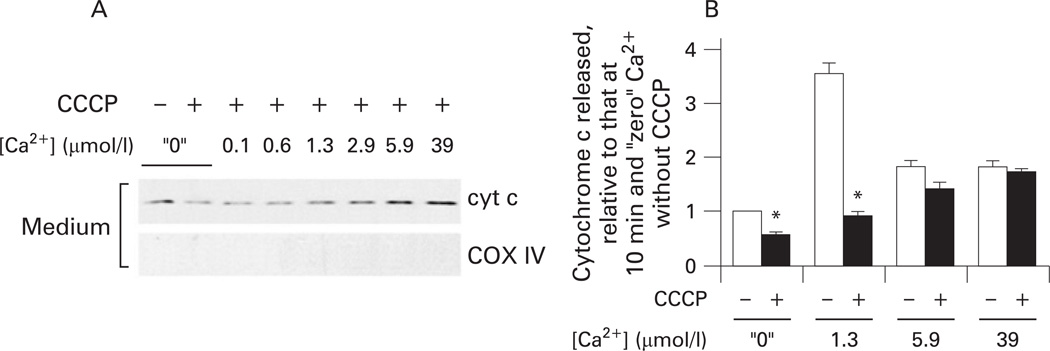
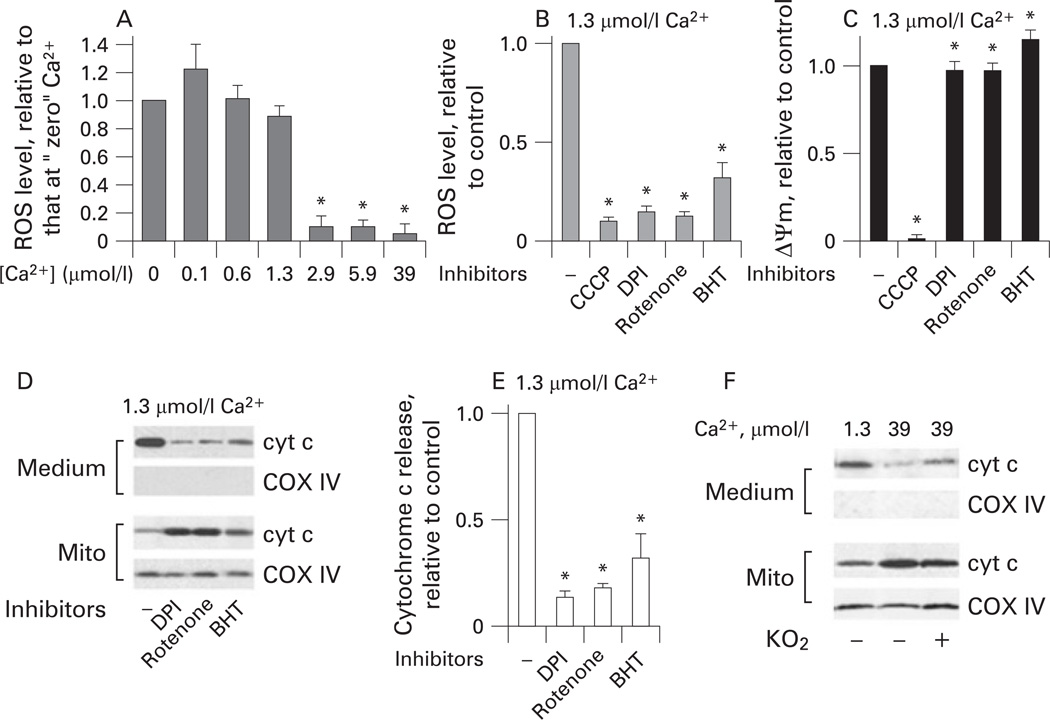
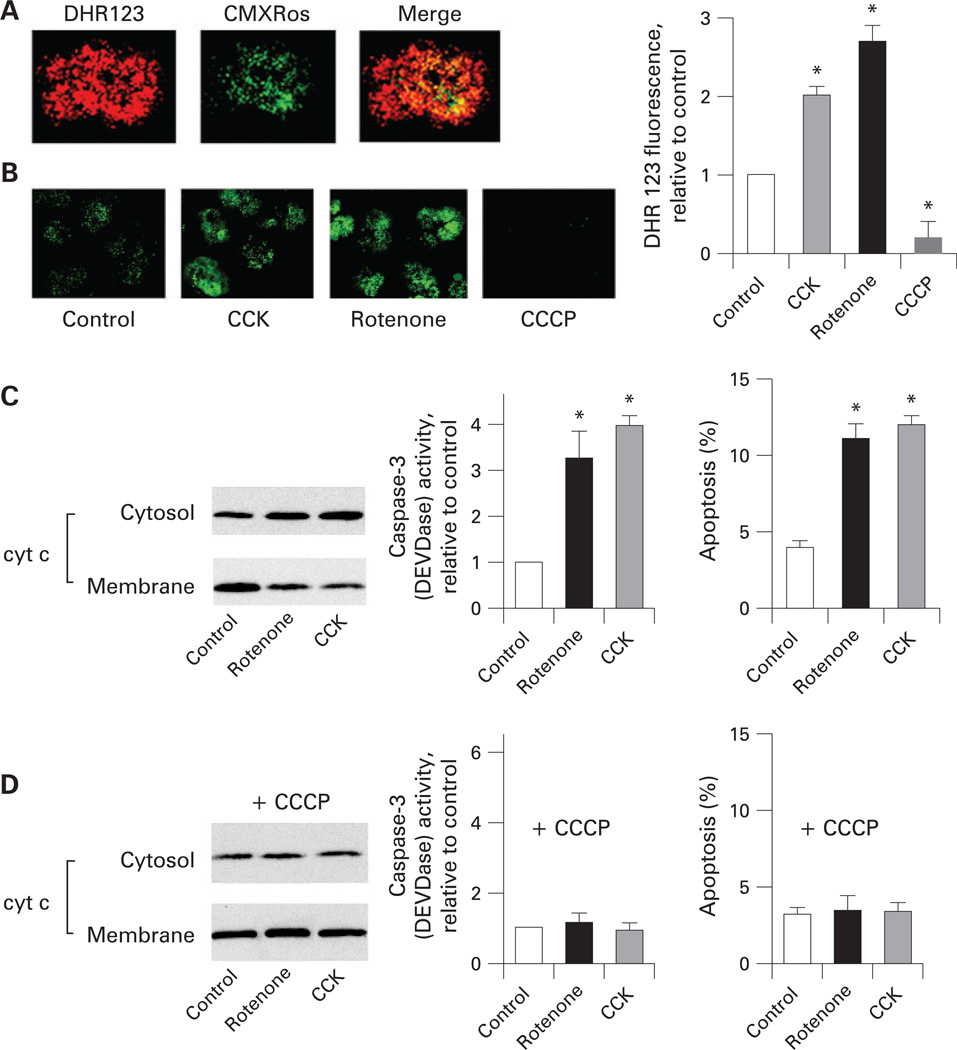
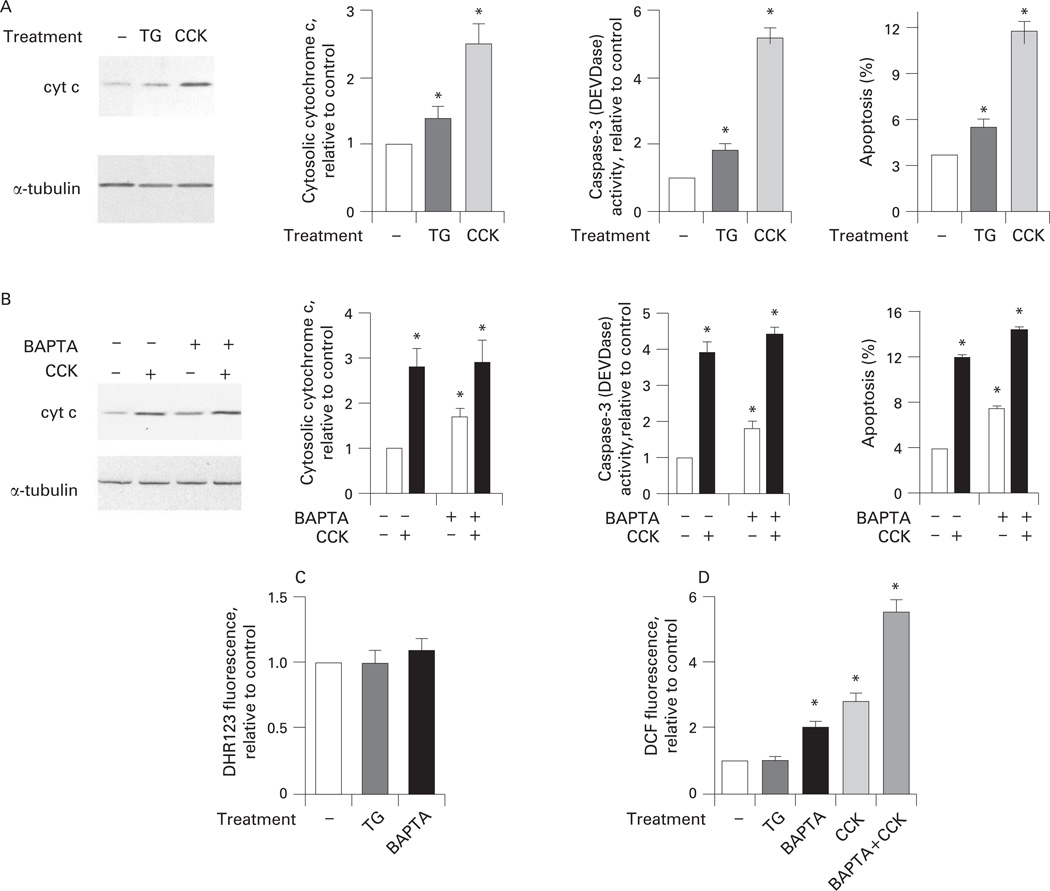
Results: Micromolar amounts of Ca(2+) depolarised isolated pancreatic mitochondria through a mechanism different from the "classical" (ie, liver) mitochondrial permeability transition pore (mPTP). In contrast with liver, Ca(2+)-induced mPTP opening caused a dramatic decrease in ROS and was not associated with pancreatic mitochondria swelling. Importantly, we found that Ca(2+)-induced depolarisation inhibited cytochrome c release from pancreatic mitochondria, due to blockade of ROS production. As a result, Ca(2+) exerted two opposite effects on cytochrome c release: Ca(2+) per se stimulated the release, whereas Ca(2+)-induced depolarisation inhibited it. This dual effect caused a non-monotonous dose-dependence of cytochrome c release on Ca(2+). In intact acinar cells, cytochrome c release, caspase activation and apoptosis were all stimulated by ROS and Ca(2+), and inhibited by depolarisation, corroborating the findings on isolated pancreatic mitochondria.
Conclusions: These data implicate ROS as a key mediator of CCK-induced apoptotic responses. The results indicate a major role for mitochondria in the effects of Ca(2+ )and ROS on acinar cell death. They suggest that the extent of apoptosis in pancreatitis is regulated by the interplay between ROS, Delta Psi m and Ca(2+). Stabilising mitochondria against loss of Delta Psi m may represent a strategy to mitigate the severity of pancreatitis.
Figures
Comment in
-
A unique pancreatic mitochondrial response to calcium and its role in apoptosis.Gut. 2009 Mar;58(3):328-30. doi: 10.1136/gut.2008.160069. Gut. 2009. PMID: 19211852 No abstract available.
References
-
- Gukovskaya AS, Pandol SJ. Cell death pathways in pancreatitis and pancreatic cancer. Pancreatology. 2004;4:567–586. - PubMed
-
- Halangk W, Lerch MM. Early events in acute pancreatitis. Clin Lab Med. 2005;25:1–15. - PubMed
-
- Kaiser AM, Saluja AK, Sengupta A, et al. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am J Physiol Cell Physiol. 1995;269:C1295–C1304. - PubMed
-
- Gukovskaya AS, Perkins P, Zaninovic V, et al. Mechanisms of cell death after pancreatic duct obstruction in the opossum and the rat. Gastroenterology. 1996;110:875–884. - PubMed
-
- Sandoval D, Gukovskaya A, Reavey P, et al. The role of neutrophils and platelet-activating factor in mediating experimental pancreatis. Gastroenterology. 1996;111:1081–1091. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous