

Update on recent molecular and genetic advances in frontotemporal lobar degeneration
- PMID: 18596549
- PMCID: PMC2761710
- DOI: 10.1097/NEN.0b013e31817d751c
Update on recent molecular and genetic advances in frontotemporal lobar degeneration
Abstract
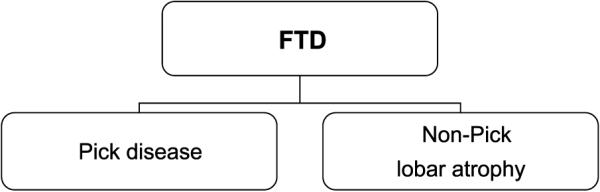
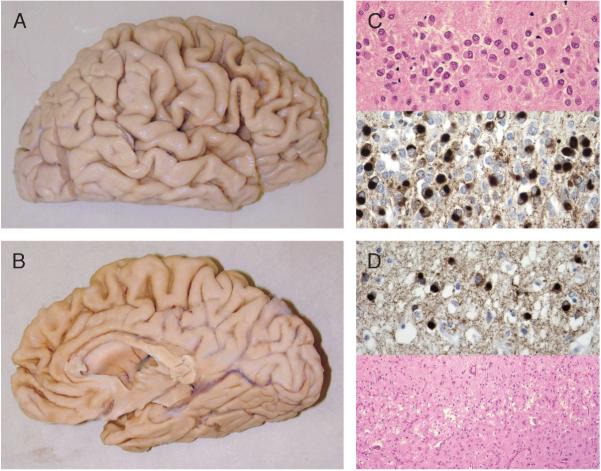
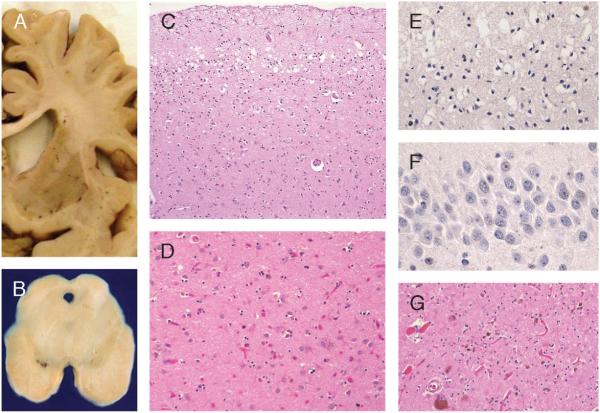
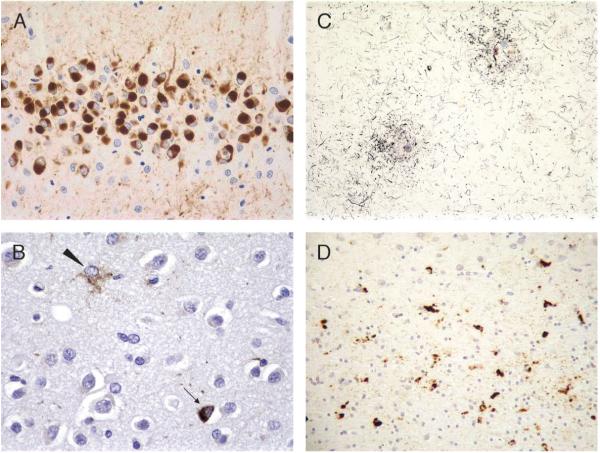
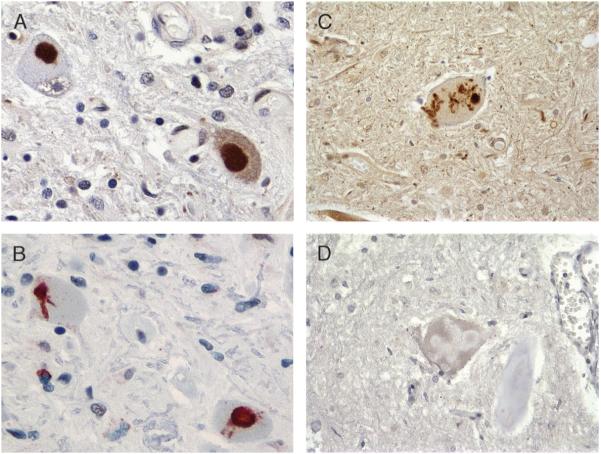
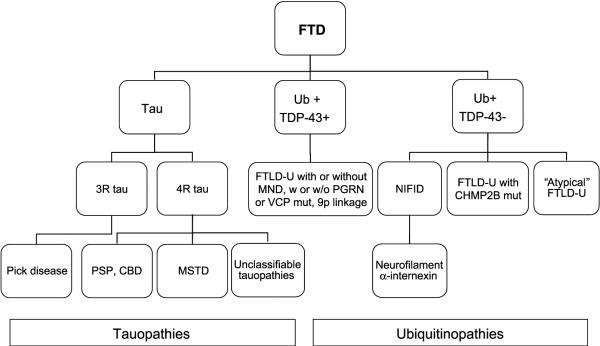
Great strides have been made in the last 2 years in the field of frontotemporal lobar degeneration (FTLD), particularly with respect to the genetics and molecular biology of FTLD with ubiquitinated inclusions. It is now clear that most cases of familial FTLD with ubiquitinated inclusions have mutations in the progranulin gene, located on chromosome 17. It is also clear that most ubiquitinated inclusions in FTLD with ubiquitinated inclusions are composed primarily of TAR DNA-binding protein-43. Thus, FTLDs can be separated into 2 major groups (i.e. tauopathies and ubiquitinopathies), and most of the ubiquitinopathies can now be defined as TAR DNA-binding protein-43 proteinopathies. Many of the familial FTLDs are linked to chromosome 17, including both the familial tauopathies and the familial TAR DNA-binding protein-43 proteinopathies with progranulin mutations. This review highlights the neuropathologic features and the most important discoveries of the last 2 years and places these findings into the historical context of FTLD.
Figures
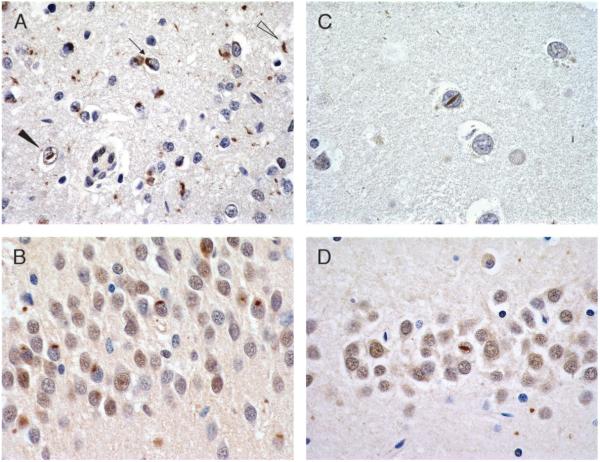
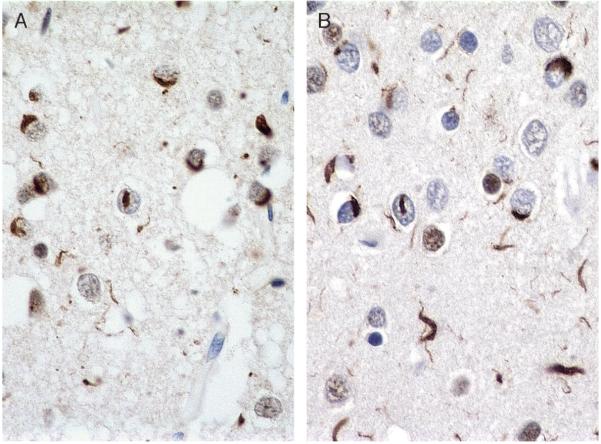
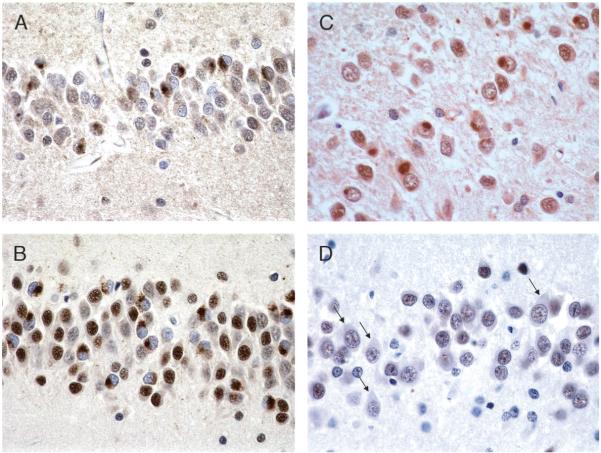
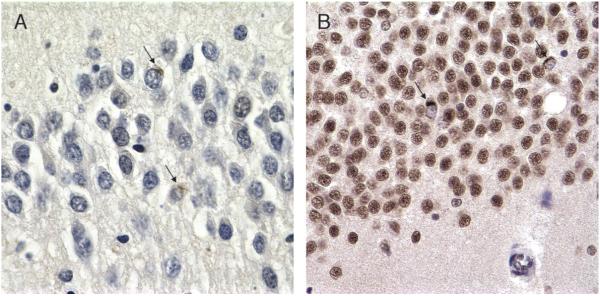
References
-
- Pick A. Über die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager Med Wochenschr. 1892;17:165–67.
-
- Alzheimer A. Über eigenartige Krankheitsfȧlle des späteren Alters. Z Gesamte Neurol Psychiat. 1911;4:356–85.
-
- Gans A. Betrachtungen über art und Ausbreitung des krankhaften Prozesses in einem Fall van Pickscher Atrophie des Stirnhins. Zeitschr f d ges Neurol u Psychiatr (Berl) 1922;80:10–28.
-
- Brun A, Gustafson L, Risberg J, et al. Frontal lobe dementia of non-Alzheimer type. Arch Gerontol Geriatr. 1987;6:193–208. - PubMed
-
- Knopman DS, Mastri AR, Frey WH, 2nd, et al. Dementia lacking distinctive histologic features: A common non-Alzheimer degenerative dementia. Neurology. 1990;40:251–56. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
