

Review
doi: 10.1172/JCI33452.
Molecular pathogenesis of pulmonary arterial hypertension
Affiliations
- PMID: 18596905
- PMCID: PMC2439479
- DOI: 10.1172/JCI33452
Item in Clipboard
Review
Molecular pathogenesis of pulmonary arterial hypertension
J Clin Invest.
2008 Jul.
Abstract
Recent investigations have suggested that it might be possible to reverse the pathology of pulmonary arterial hypertension (PAH), a disorder that can be rapidly progressive and fatal despite current treatments including i.v. prostacyclin. This review will address the cellular and molecular processes implicated in clinical, genetic, and experimental studies as underlying the pulmonary vascular abnormalities associated with PAH. Emerging treatments are aimed at inducing apoptosis of abnormal vascular cells that obstruct blood flow and at promoting regeneration of "lost" distal vasculature.
Figures
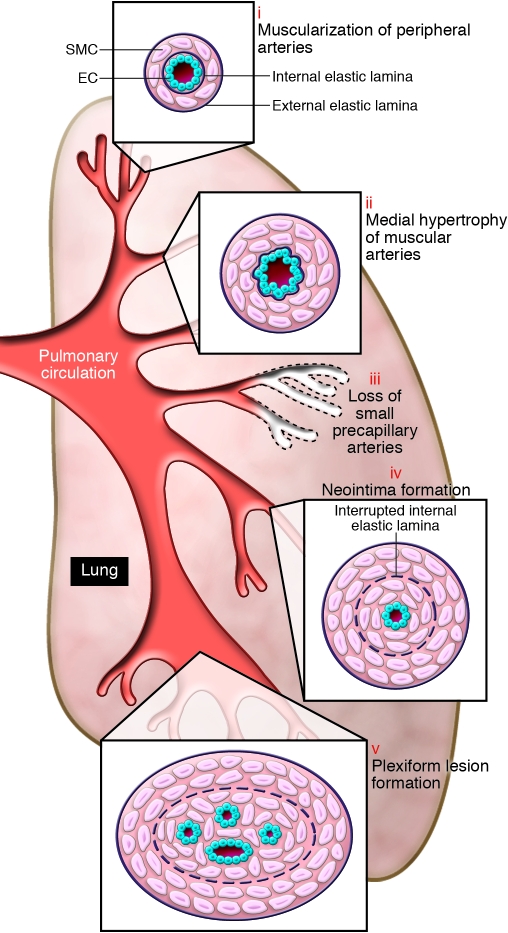
Schema illustrating the different vascular abnormalities compared with normal pulmonary circulation, associated with PH. This schema depicts the abnormalities throughout the pulmonary circulation, including (i) abnormal muscularization of distal precapillary arteries, (ii) medial hypertrophy (thickening) of large pulmonary muscular arteries, (iii) loss of precapillary arteries, (iv) neointimal formation that is particularly occlusive in vessels 100–500 μM, and (v) formation of plexiform lesions in these vessels.
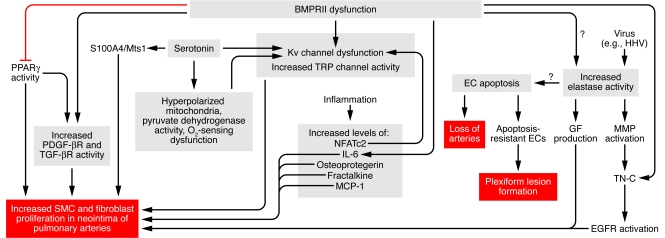
This schema focuses on factors causing increased SMC and fibroblast proliferation as well as apoptosis of ECs, causing an initial reduction in vessel number, followed by proliferation of apoptosis-resistant ECs in plexiform lesions. It shows multiple levels of interaction, with numerous factors related as described in the text. For example, serotonin stimulates both PDGF-mediated and S100A4/Mts1-mediated SMC and fibroblast proliferation and it also reduces Kv channel function, as does hyperpolarized mitochondria. BMPRII dysfunction causes Kv channel dysfunction and enhances TRP channel activity, which increases intracellular calcium levels, and may (as reflected by question mark) induce elastase activity. Viruses of the herpes family can induce elastase activity. Elastase, via activation of MMPs and tenascin C (TN-C), upregulates growth factor (GF) receptors such as EGF receptors (EGFRs) and also triggers release of growth factors such as EGF from the extracellular matrix, all of which leads to SMC proliferation. BMPRII dysfunction can also increase PDGF activity, increase SMC proliferation by suppressing PPARγ, and increase TGF-β activity. BMPRII dysfunction can enhance inflammation via osteoprotegrin and IL-6. NFATc2 can suppress Kv channel function. Other inflammatory mediators such as fractalkine and MCP-1 can, in addition to osteoprotegrin and IL-6, increase SMC proliferation. BMPRII dysfunction can lead to EC apoptosis, as can elastase activity. EC apoptosis may predispose to the development of apoptosis-resistant ECs in plexiform lesions. MCP-1, mast cell proteinase 1; TRP, transient receptor potential Ca2+ channels.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
