

Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations
- PMID: 18596924
- PMCID: PMC2441858
- DOI: 10.1172/JCI35414
Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations
Abstract
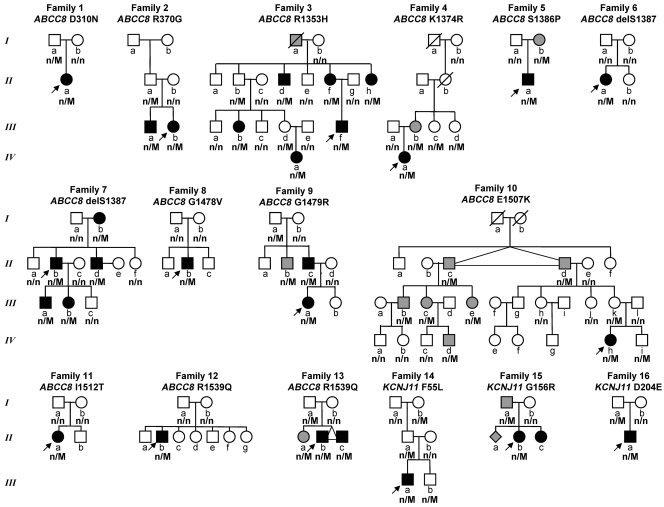
Congenital hyperinsulinism is a condition of dysregulated insulin secretion often caused by inactivating mutations of the ATP-sensitive K+ (KATP) channel in the pancreatic beta cell. Though most disease-causing mutations of the 2 genes encoding KATP subunits, ABCC8 (SUR1) and KCNJ11 (Kir6.2), are recessively inherited, some cases of dominantly inherited inactivating mutations have been reported. To better understand the differences between dominantly and recessively inherited inactivating KATP mutations, we have identified and characterized 16 families with 14 different dominantly inherited KATP mutations, including a total of 33 affected individuals. The 16 probands presented with hypoglycemia at ages from birth to 3.3 years, and 15 of 16 were well controlled on diazoxide, a KATP channel agonist. Of 29 adults with mutations, 14 were asymptomatic. In contrast to a previous report of increased diabetes risk in dominant KATP hyperinsulinism, only 4 of 29 adults had diabetes. Unlike recessive mutations, dominantly inherited KATP mutant subunits trafficked normally to the plasma membrane when expressed in COSm6 cells. Dominant mutations also resulted in different channel-gating defects, as dominant ABCC8 mutations diminished channel responses to magnesium adenosine diphosphate or diazoxide, while dominant KCNJ11 mutations impaired channel opening, even in the absence of nucleotides. These data highlight distinctive features of dominant KATP hyperinsulinism relative to the more common and more severe recessive form, including retention of normal subunit trafficking, impaired channel activity, and a milder hypoglycemia phenotype that may escape detection in infancy and is often responsive to diazoxide medical therapy, without the need for surgical pancreatectomy.
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
