

Age-dependent demise of GNAS-mutated skeletal stem cells and "normalization" of fibrous dysplasia of bone
- PMID: 18597624
- PMCID: PMC2585500
- DOI: 10.1359/jbmr.080609
Age-dependent demise of GNAS-mutated skeletal stem cells and "normalization" of fibrous dysplasia of bone
Abstract
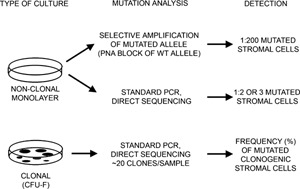
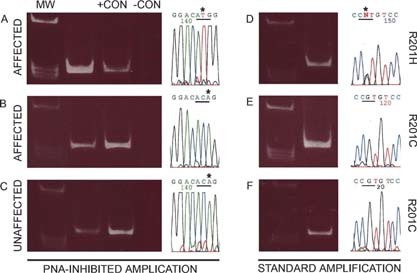
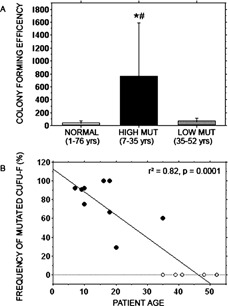
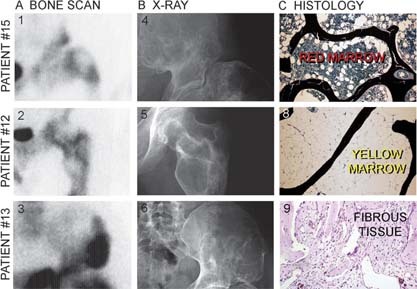
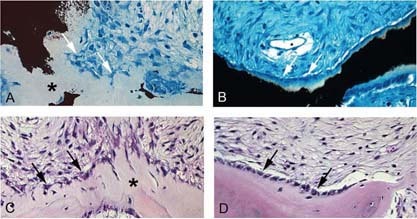
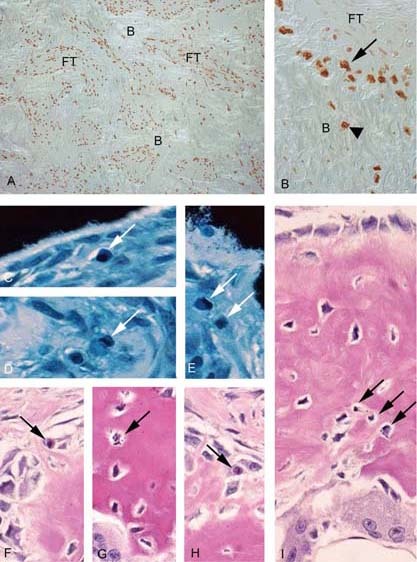
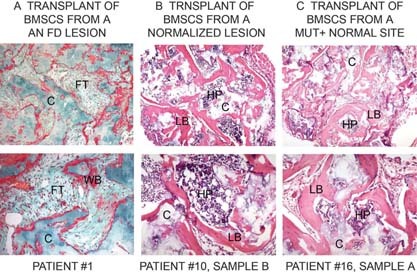
We studied the role of somatic mosaicism in fibrous dysplasia of bone (FD) within the context of skeletal ("mesenchymal") stem cells by assessing the frequency of mutated colony forming unit-fibroblasts (CFU-Fs) from FD lesions, and in some cases, from unaffected sites, in a series of patients. There was a tight inverse correlation between the percentage mutant CFU-F versus age, suggesting demise of mutant stem cells caused by exuberant apoptosis noted in samples from young patients. In older patients, either partially or completely normal bone/marrow histology was observed. On in vivo transplantation, FD ossicles were generated only by cell strains in which mutant CFU-Fs were identified. Strains that lacked mutant CFU-F (but were mutation positive) failed to regenerate an FD ossicle. These data indicate that GNAS mutations are only pathogenic when in clonogenic skeletal stem cells. From these data, we have evolved the novel concept of "normalization" of FD. As a lesion ages, mutant stem cells fail to self-renew, and their progeny are consumed by apoptosis, whereas residual normal stem cells survive, self-renew, and enable formation of a normal structure. This suggests that activating GNAS mutations disrupt a pathway that is required for skeletal stem cell self-renewal.
Figures
Similar articles
-
Direct evidence for the age-dependent demise of GNAS-mutated cells in oral fibrous dysplasia.Arch Oral Biol. 2018 Sep;93:133-140. doi: 10.1016/j.archoralbio.2018.05.018. Epub 2018 May 30. Arch Oral Biol. 2018. PMID: 29909118
-
GNAS transcripts in skeletal progenitors: evidence for random asymmetric allelic expression of Gs alpha.Hum Mol Genet. 2007 Aug 15;16(16):1921-30. doi: 10.1093/hmg/ddm139. Epub 2007 Jun 12. Hum Mol Genet. 2007. PMID: 17566083
-
Mutations of the GNAS1 gene, stromal cell dysfunction, and osteomalacic changes in non-McCune-Albright fibrous dysplasia of bone.J Bone Miner Res. 2000 Jan;15(1):120-8. doi: 10.1359/jbmr.2000.15.1.120. J Bone Miner Res. 2000. PMID: 10646121
-
Skeletal progenitors and the GNAS gene: fibrous dysplasia of bone read through stem cells.J Mol Endocrinol. 2010 Dec;45(6):355-64. doi: 10.1677/JME-10-0097. Epub 2010 Sep 14. J Mol Endocrinol. 2010. PMID: 20841428 Free PMC article. Review.
-
Fibrous Dysplasia/McCune-Albright Syndrome: Clinical and Translational Perspectives.Curr Osteoporos Rep. 2016 Oct;14(5):178-86. doi: 10.1007/s11914-016-0317-0. Curr Osteoporos Rep. 2016. PMID: 27492469 Free PMC article. Review.
Cited by
-
Fibrous dysplasia for radiologists: beyond ground glass bone matrix.Insights Imaging. 2018 Dec;9(6):1035-1056. doi: 10.1007/s13244-018-0666-6. Epub 2018 Nov 27. Insights Imaging. 2018. PMID: 30484079 Free PMC article. Review.
-
Patient-Derived Organoids Recapitulate Pathological Intrinsic and Phenotypic Features of Fibrous Dysplasia.Cells. 2024 Apr 23;13(9):729. doi: 10.3390/cells13090729. Cells. 2024. PMID: 38727265 Free PMC article.
-
Cutaneous skeletal hypophosphatemia syndrome: clinical spectrum, natural history, and treatment.Osteoporos Int. 2016 Dec;27(12):3615-3626. doi: 10.1007/s00198-016-3702-8. Epub 2016 Aug 6. Osteoporos Int. 2016. PMID: 27497815 Free PMC article. Review.
-
[Fibrous dysplasia].Orthopadie (Heidelb). 2024 Oct;53(10):805-816. doi: 10.1007/s00132-024-04548-w. Epub 2024 Sep 5. Orthopadie (Heidelb). 2024. PMID: 39235640 German.
-
Quantitative analysis of activating alpha subunit of the G protein (Gsα) mutation by pyrosequencing in fibrous dysplasia and other bone lesions.J Mol Diagn. 2011 Mar;13(2):137-42. doi: 10.1016/j.jmoldx.2010.10.003. J Mol Diagn. 2011. PMID: 21354047 Free PMC article.
References
-
- Bianco P, Riminucci M, Majolagbe A, Kuznetsov SA, Collins MT, Mankani MH, Corsi A, Bone HG, Wientroub S, Spiegel AM, Fisher LW, Robey PG 2000. Mutations of the GNAS1 gene, stromal cell dysfunction, and osteomalacic changes in non‐McCune‐Albright fibrous dysplasia of bone. J Bone Miner Res 15: 120–128. - PubMed
-
- Albright F, Butler AM, Hampton AO, Smith P 1937. Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction with precocious puberty in females. N Engl J Med 216: 727–746.
-
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM 1991. Activating mutations of the stimulatory G protein in the McCune‐Albright syndrome. N Engl J Med 325: 1688–1695. - PubMed
-
- Shenker A, Weinstein LS, Sweet DE, Spiegel AM 1994. An activating Gs alpha mutation is present in fibrous dysplasia of bone in the McCune‐Albright syndrome. J Clin Endocrinol Metab 79: 750–755. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
