

High accuracy mass spectrometry analysis as a tool to verify and improve gene annotation using Mycobacterium tuberculosis as an example
- PMID: 18597682
- PMCID: PMC2483986
- DOI: 10.1186/1471-2164-9-316
High accuracy mass spectrometry analysis as a tool to verify and improve gene annotation using Mycobacterium tuberculosis as an example
Abstract
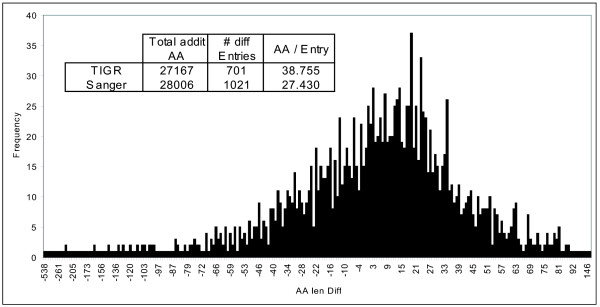
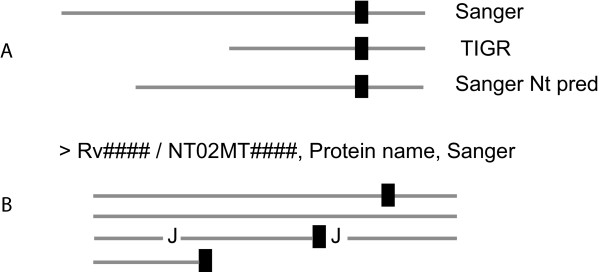
Background: While the genomic annotations of diverse lineages of the Mycobacterium tuberculosis complex are available, divergences between gene prediction methods are still a challenge for unbiased protein dataset generation. M. tuberculosis gene annotation is an example, where the most used datasets from two independent institutions (Sanger Institute and Institute of Genomic Research-TIGR) differ up to 12% in the number of annotated open reading frames, and 46% of the genes contained in both annotations have different start codons. Such differences emphasize the importance of the identification of the sequence of protein products to validate each gene annotation including its sequence coding area.
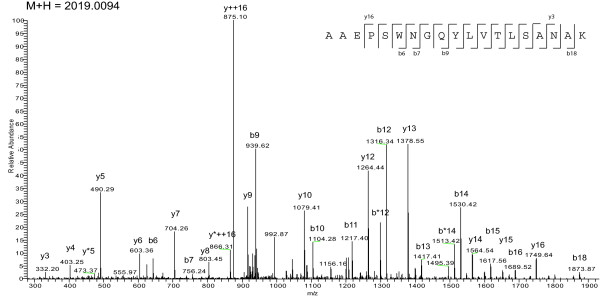
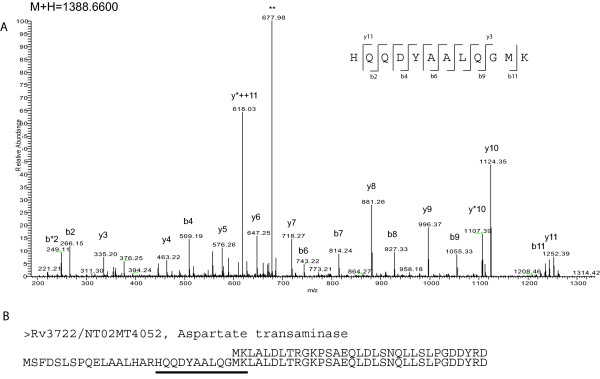
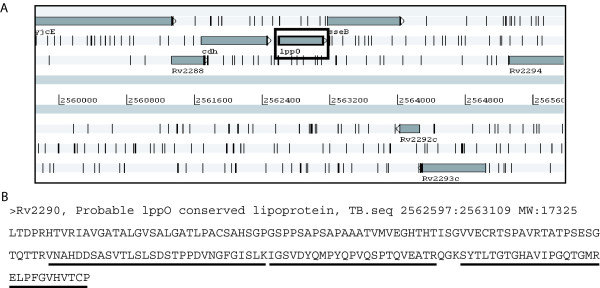
Results: With this objective, we submitted a culture filtrate sample from M. tuberculosis to a high-accuracy LTQ-Orbitrap mass spectrometer analysis and applied refined N-terminal prediction to perform comparison of two gene annotations. From a total of 449 proteins identified from the MS data, we validated 35 tryptic peptides that were specific to one of the two datasets, representing 24 different proteins. From those, 5 proteins were only annotated in the Sanger database. In the remaining proteins, the observed differences were due to differences in annotation of transcriptional start sites.
Conclusion: Our results indicate that, even in a less complex sample likely to represent only 10% of the bacterial proteome, we were still able to detect major differences between different gene annotation approaches. This gives hope that high-throughput proteomics techniques can be used to improve and validate gene annotations, and in particular for verification of high-throughput, automatic gene annotations.
Figures

References
-
- World Health Organization. WHO Report 2007: Global tuberculosis control, surveillance, planning, financing. 2007.
-
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. - DOI - PubMed
-
- Eiglmeier K, Simon S, Garnier T, Cole ST. The integrated genome map of Mycobacterium leprae. Leprosy review. 2001;72:462–469. - PubMed
-
- Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, Peterson J, DeBoy R, Dodson R, Gwinn M, Haft D, Hickey E, Kolonay JF, Nelson WC, Umayam LA, Ermolaeva M, Salzberg SL, Delcher A, Utterback T, Weidman J, Khouri H, Gill J, Mikula A, Bishai W, Jacobs Jr WR, Jr., Venter JC, Fraser CM. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. Journal of bacteriology. 2002;184:5479–5490. doi: 10.1128/JB.184.19.5479-5490.2002. - DOI - PMC - PubMed
-
- Garnier T, Eiglmeier K, Camus JC, Medina N, Mansoor H, Pryor M, Duthoy S, Grondin S, Lacroix C, Monsempe C, Simon S, Harris B, Atkin R, Doggett J, Mayes R, Keating L, Wheeler PR, Parkhill J, Barrell BG, Cole ST, Gordon SV, Hewinson RG. The complete genome sequence of Mycobacterium bovis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7877–7882. doi: 10.1073/pnas.1130426100. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
