

TDAG51 is an ERK signaling target that opposes ERK-mediated HME16C mammary epithelial cell transformation
- PMID: 18597688
- PMCID: PMC2474852
- DOI: 10.1186/1471-2407-8-189
TDAG51 is an ERK signaling target that opposes ERK-mediated HME16C mammary epithelial cell transformation
Abstract
Introduction: Signaling downstream of Ras is mediated by three major pathways, Raf/ERK, phosphatidylinositol 3 kinase (PI3K), and Ral guanine nucleotide exchange factor (RalGEF). Ras signal transduction pathways play an important role in breast cancer progression, as evidenced by the frequent over-expression of the Ras-activating epidermal growth factor receptors EGFR and ErbB2. Here we investigated which signal transduction pathways downstream of Ras contribute to EGFR-dependent transformation of telomerase-immortalized mammary epithelial cells HME16C. Furthermore, we examined whether a highly transcriptionally regulated ERK pathway target, PHLDA1 (TDAG51), suggested to be a tumor suppressor in breast cancer and melanoma, might modulate the transformation process.
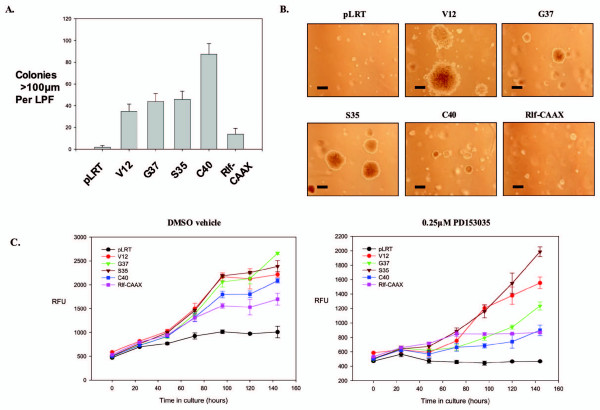
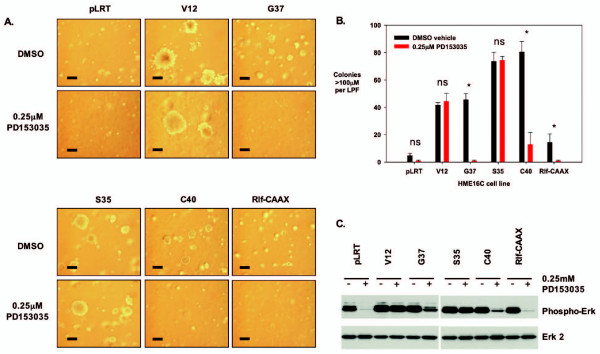
Methods: Cellular transformation of human mammary epithelial cells by downstream Ras signal transduction pathways was examined using anchorage-independent growth assays in the presence and absence of EGFR inhibition. TDAG51 protein expression was down-regulated by interfering small hairpin RNA (shRNA), and the effects on cell proliferation and death were examined in Ras pathway-transformed breast epithelial cells.
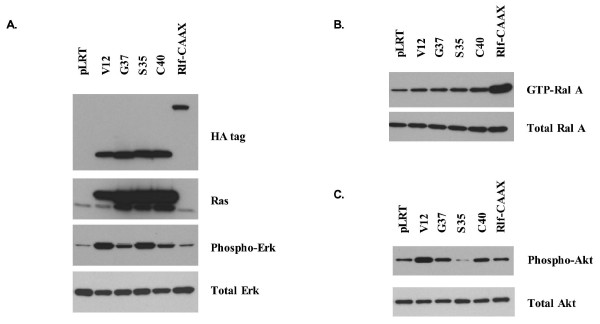
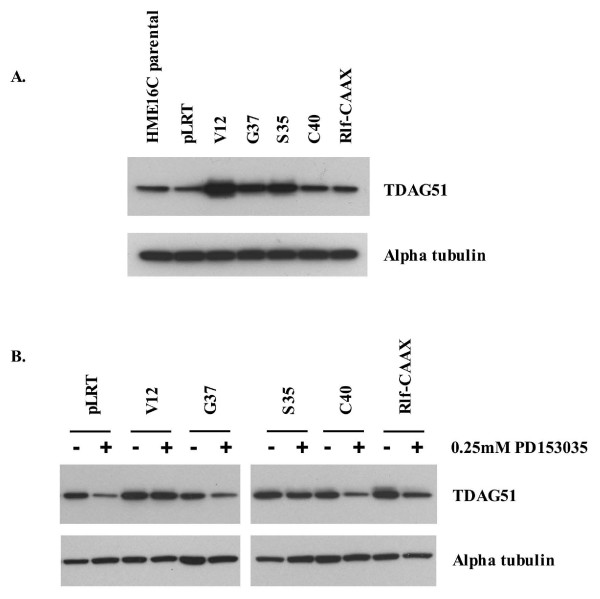
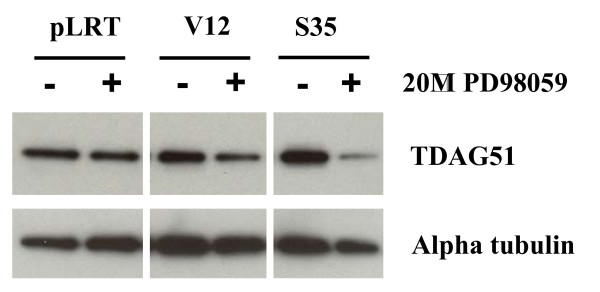
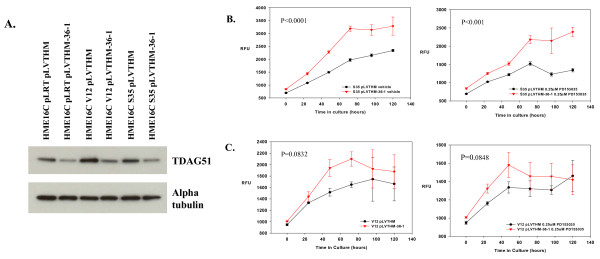
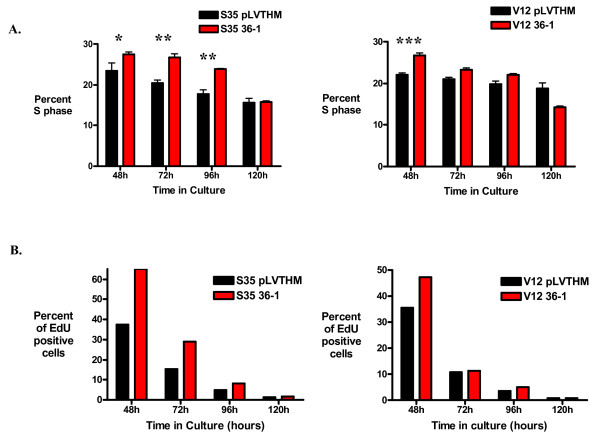
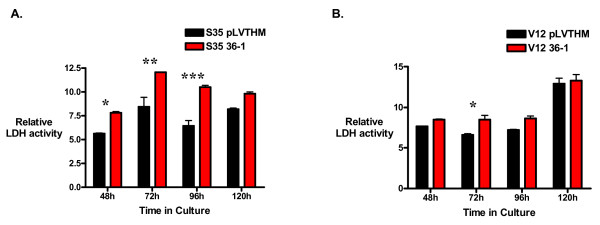
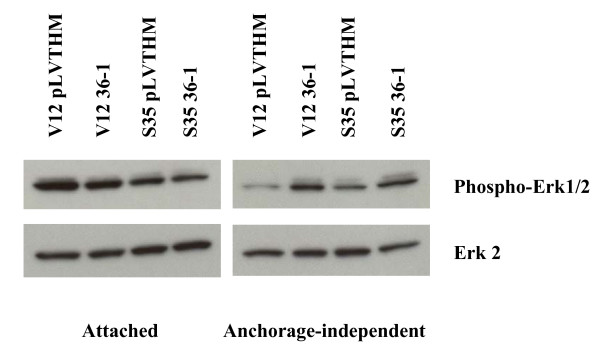
Results: Activation of both the ERK and PI3K signaling pathways was sufficient to induce cellular transformation, which was accompanied by up-regulation of EGFR ligands, suggesting autocrine EGFR stimulation during the transformation process. Only activation of the ERK pathway was sufficient to transform cells in the presence of EGFR inhibition and was sufficient for tumorigenesis in xenografts. Up-regulation of the PHLDA1 gene product, TDAG51, was found to correlate with persistent ERK activation and anchorage-independent growth in the absence or presence of EGFR inhibition. Knockdown of this putative breast cancer tumor-suppressor gene resulted in increased ERK pathway activation and enhanced matrix-detached cellular proliferation of Ras/Raf transformed cells.
Conclusion: Our results suggest that multiple Ras signal transduction pathways contribute to mammary epithelial cell transformation, but that the ERK signaling pathway may be a crucial component downstream of EGFR activation during tumorigenesis. Furthermore, persistent activation of ERK signaling up-regulates TDAG51. This event serves as a negative regulator of both Erk activation as well as matrix-detached cellular proliferation and suggests that TDAG51 opposes ERK-mediated transformation in breast epithelial cells.
Figures
References
-
- Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. - PubMed
-
- Mueller H, Flury N, Eppenberger-Castori S, Kueng W, David F, Eppenberger U. Potential prognostic value of mitogen-activated protein kinase activity for disease-free survival of primary breast cancer patients. Int J Cancer. 2000;89:384–388. doi: 10.1002/1097-0215(20000720)89:4<384::AID-IJC11>3.0.CO;2-R. - DOI - PubMed
-
- Abd El-Rehim DM, Pinder SE, Paish CE, Bell JA, Rampaul RS, Blamey RW, Robertson JF, Nicholson RI, Ellis IO. Expression and co-expression of the members of the epidermal growth factor receptor (EGFR) family in invasive breast carcinoma. Br J Cancer. 2004;91:1532–1542. doi: 10.1038/sj.bjc.6602184. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
