

Seeded Bayesian Networks: constructing genetic networks from microarray data
- PMID: 18601736
- PMCID: PMC2474592
- DOI: 10.1186/1752-0509-2-57
Seeded Bayesian Networks: constructing genetic networks from microarray data
Abstract
Background: DNA microarrays and other genomics-inspired technologies provide large datasets that often include hidden patterns of correlation between genes reflecting the complex processes that underlie cellular metabolism and physiology. The challenge in analyzing large-scale expression data has been to extract biologically meaningful inferences regarding these processes - often represented as networks - in an environment where the datasets are often imperfect and biological noise can obscure the actual signal. Although many techniques have been developed in an attempt to address these issues, to date their ability to extract meaningful and predictive network relationships has been limited. Here we describe a method that draws on prior information about gene-gene interactions to infer biologically relevant pathways from microarray data. Our approach consists of using preliminary networks derived from the literature and/or protein-protein interaction data as seeds for a Bayesian network analysis of microarray results.
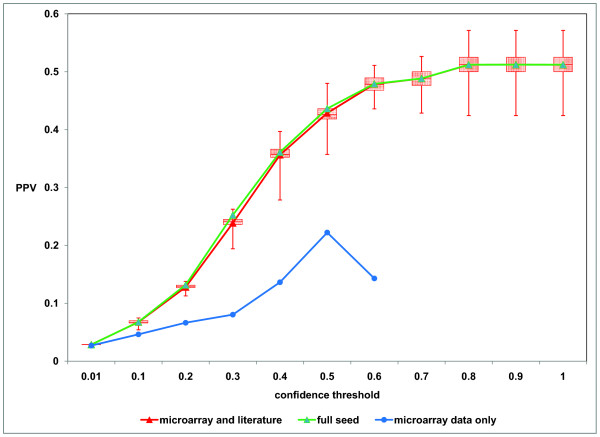
Results: Through a bootstrap analysis of gene expression data derived from a number of leukemia studies, we demonstrate that seeded Bayesian Networks have the ability to identify high-confidence gene-gene interactions which can then be validated by comparison to other sources of pathway data.
Conclusion: The use of network seeds greatly improves the ability of Bayesian Network analysis to learn gene interaction networks from gene expression data. We demonstrate that the use of seeds derived from the biomedical literature or high-throughput protein-protein interaction data, or the combination, provides improvement over a standard Bayesian Network analysis, allowing networks involving dynamic processes to be deduced from the static snapshots of biological systems that represent the most common source of microarray data. Software implementing these methods has been included in the widely used TM4 microarray analysis package.
Figures
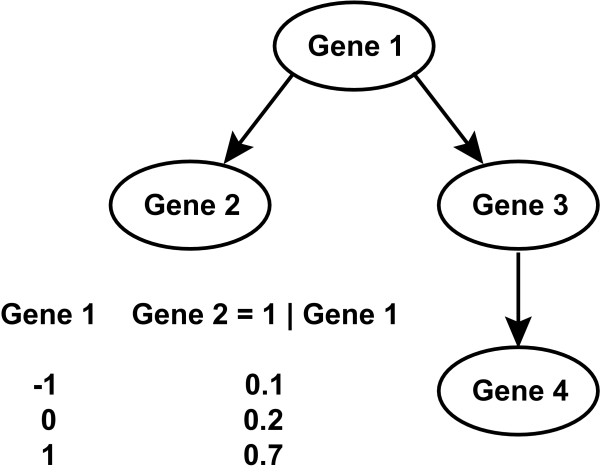
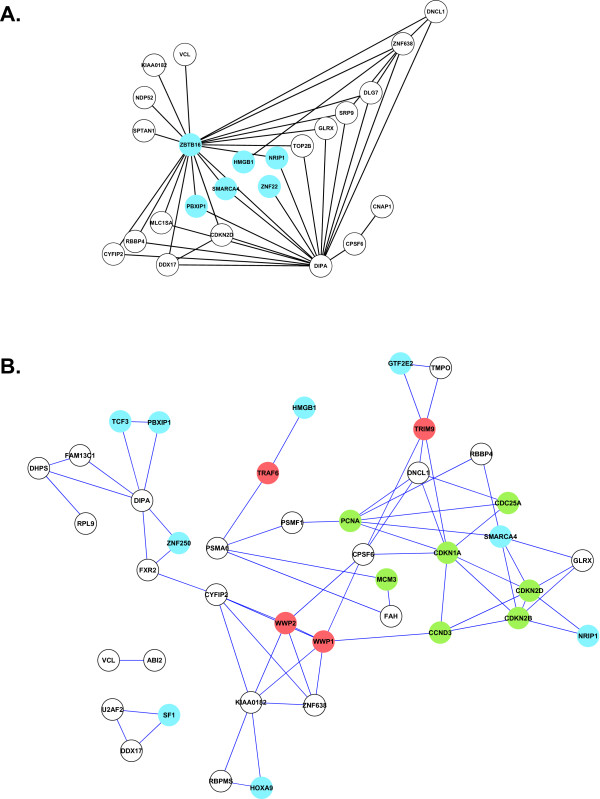
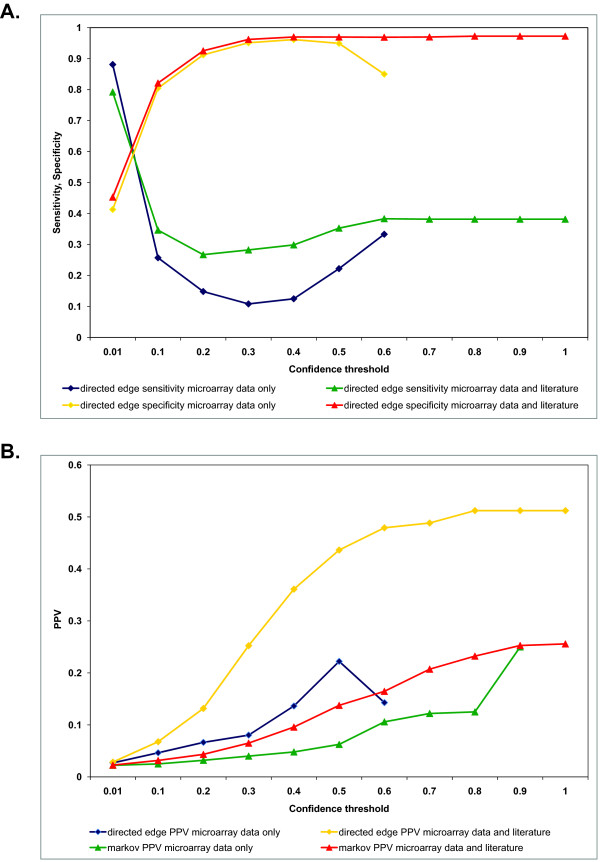
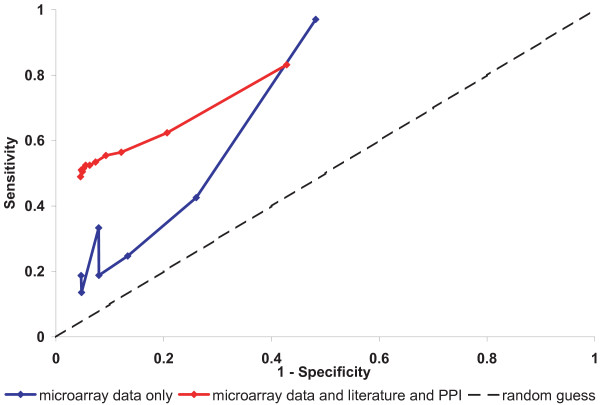
References
-
- Weaver DC, Workman CT, Stormo GD. Modeling regulatory networks with weight matrices. Pac Symp Biocomput. 1999:112–123. - PubMed
-
- Akutsu T, Miyano S, Kuhara S. Identification of genetic networks from a small number of gene expression patterns under the Boolean network model. Pac Symp Biocomput. 1999:17–28. - PubMed
-
- Chen T, He HL, Church GM. Modeling gene expression with differential equations. Pac Symp Biocomput. 1999:29–40. - PubMed
-
- Friedman N, Linial M, Nachman I, Pe'er D. Using Bayesian networks to analyze expression data. J Comput Biol. 2000;7:601–620. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
