

Congenital long QT syndrome
- PMID: 18606002
- PMCID: PMC2474834
- DOI: 10.1186/1750-1172-3-18
Congenital long QT syndrome
Abstract
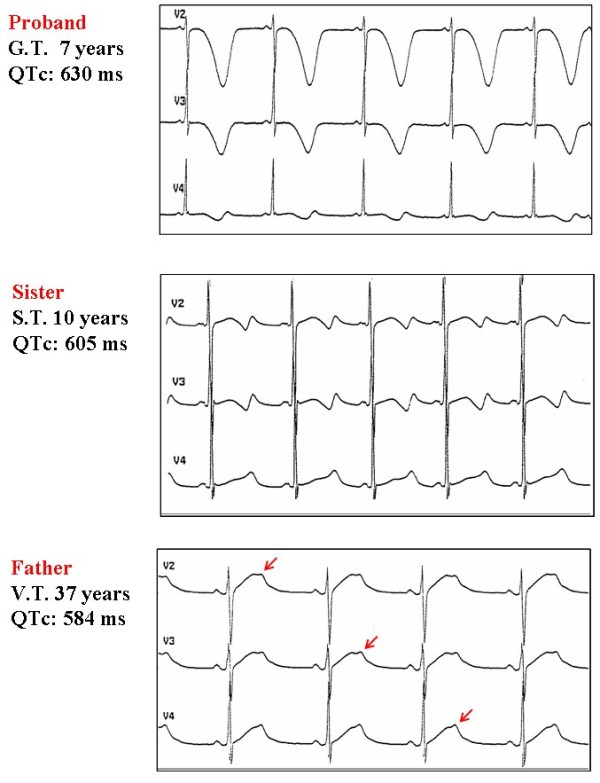
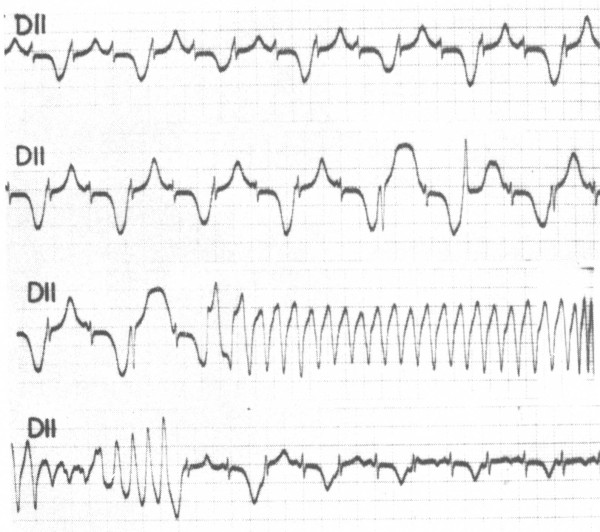
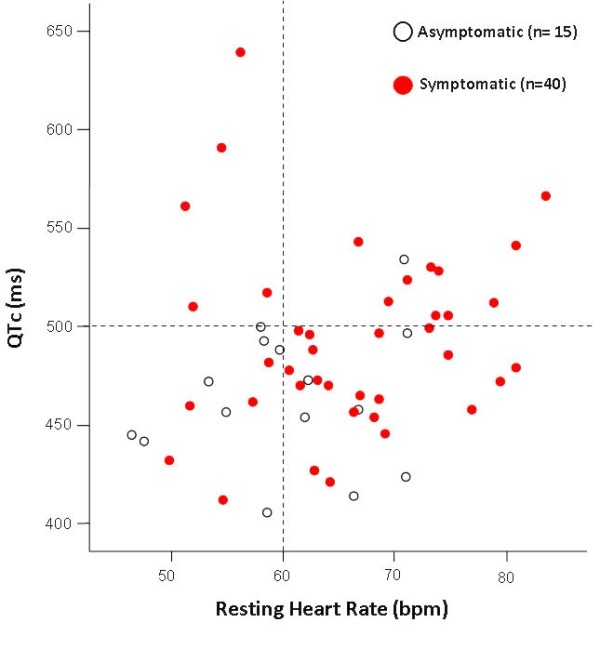
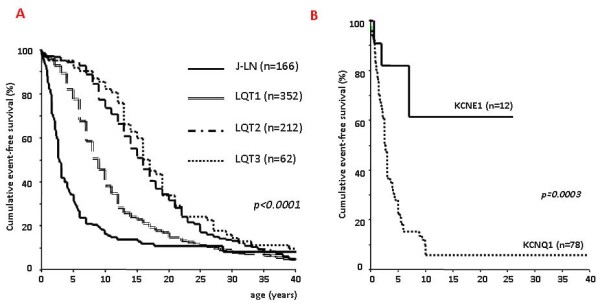
Congenital long QT syndrome (LQTS) is a hereditary cardiac disease characterized by a prolongation of the QT interval at basal ECG and by a high risk of life-threatening arrhythmias. Disease prevalence is estimated at close to 1 in 2,500 live births. The two cardinal manifestations of LQTS are syncopal episodes, that may lead to cardiac arrest and sudden cardiac death, and electrocardiographic abnormalities, including prolongation of the QT interval and T wave abnormalities. The genetic basis of the disease was identified in the mid-nineties and all the LQTS genes identified so far encode cardiac ion channel subunits or proteins involved in modulating ionic currents. Mutations in these genes (KCNQ1, KCNH2, KCNE1, KCNE2, CACNA1c, CAV3, SCN5A, SCN4B) cause the disease by prolonging the duration of the action potential. The most prevalent LQTS variant (LQT1) is caused by mutations in the KCNQ1 gene, with approximately half of the genotyped patients carrying KCNQ1 mutations. Given the characteristic features of LQTS, the typical cases present no diagnostic difficulties for physicians aware of the disease. However, borderline cases are more complex and require the evaluation of various electrocardiographic, clinical, and familial findings, as proposed in specific diagnostic criteria. Additionally, molecular screening is now part of the diagnostic process. Treatment should always begin with beta-blockers, unless there are valid contraindications. If the patient has one more syncope despite a full dose beta-blockade, left cardiac sympathetic denervation (LCSD) should be performed without hesitation and implantable cardioverter defibrillator (ICD) therapy should be considered with the final decision being based on the individual patient characteristics (age, sex, clinical history, genetic subgroup including mutation-specific features in some cases, presence of ECG signs - including 24-hour Holter recordings - indicating high electrical instability). The prognosis of the disease is usually good in patients that are correctly diagnosed and treated. However, there are a few exceptions: patients with Timothy syndrome, patients with Jervell Lange-Nielsen syndrome carrying KCNQ1 mutations and LQT3 patients with 2:1 atrio-ventricular block and very early occurrence of cardiac arrhythmias.
Figures
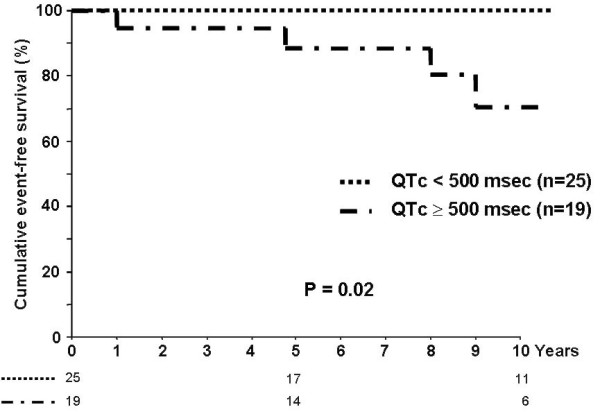
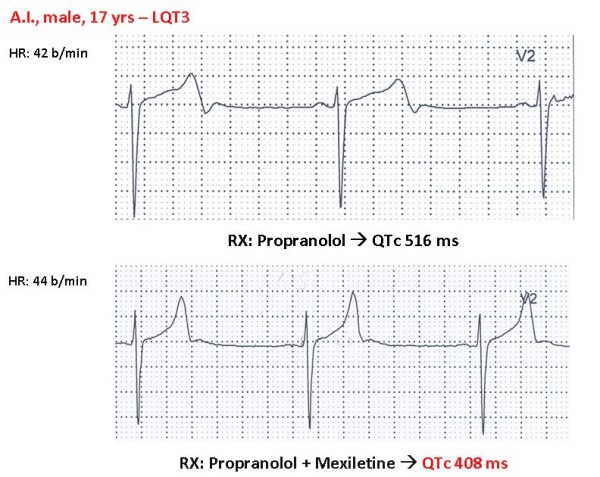
References
-
- Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, Shkolnikova M, Berul CI, Bitner-Glindzicz M, Toivonen L, Horie M, Schulze-Bahr E, Denjoy I. The Jervell and Lange-Nielsen Syndrome. Natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783–790. doi: 10.1161/CIRCULATIONAHA.105.592899. - DOI - PubMed
-
- Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare dell'età pediatrica. Clin Pediat. 1963;45:656–683. - PubMed
-
- Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103–106. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
