

Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding
- PMID: 18606141
- PMCID: PMC2586299
- DOI: 10.1016/j.devcel.2008.05.014
Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding
Abstract
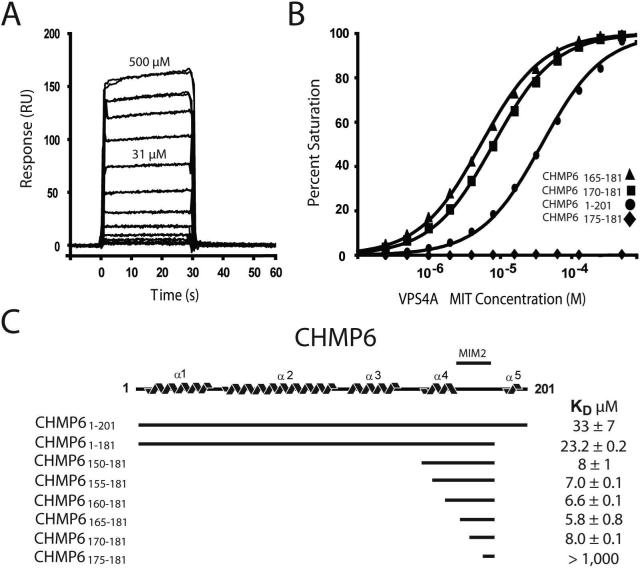
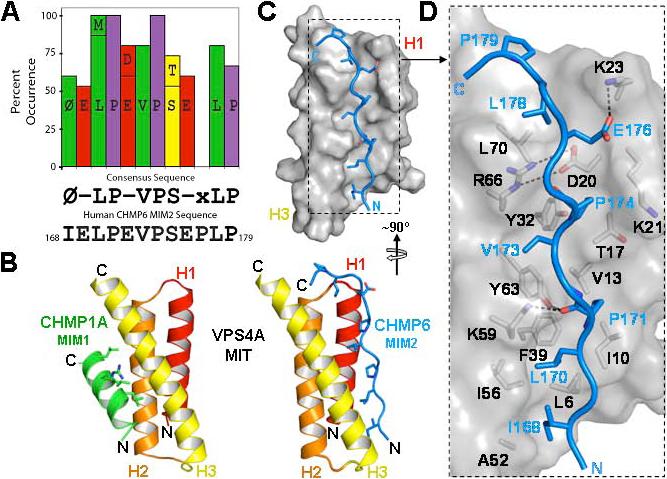
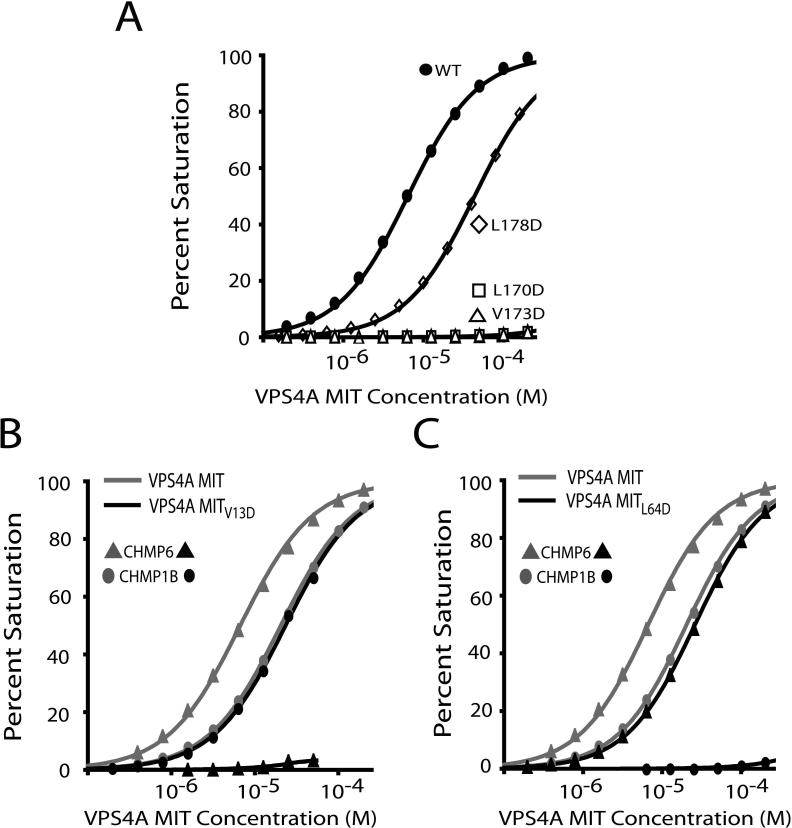
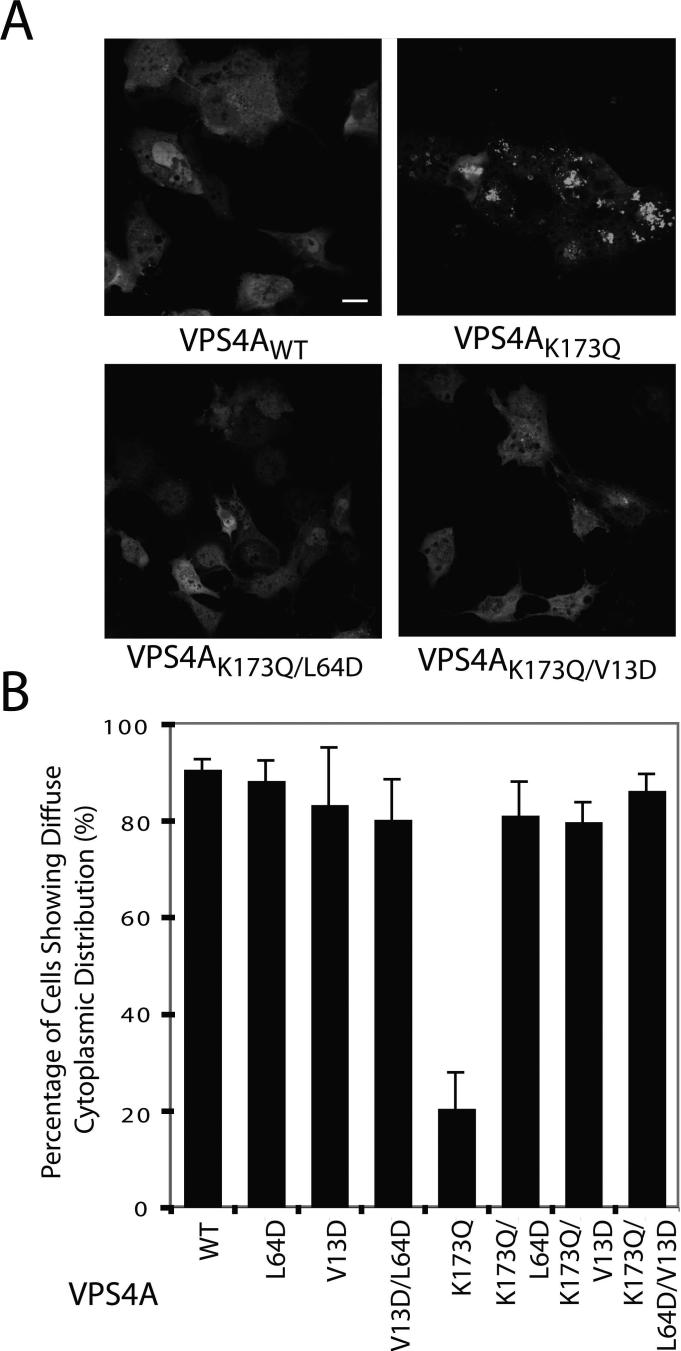
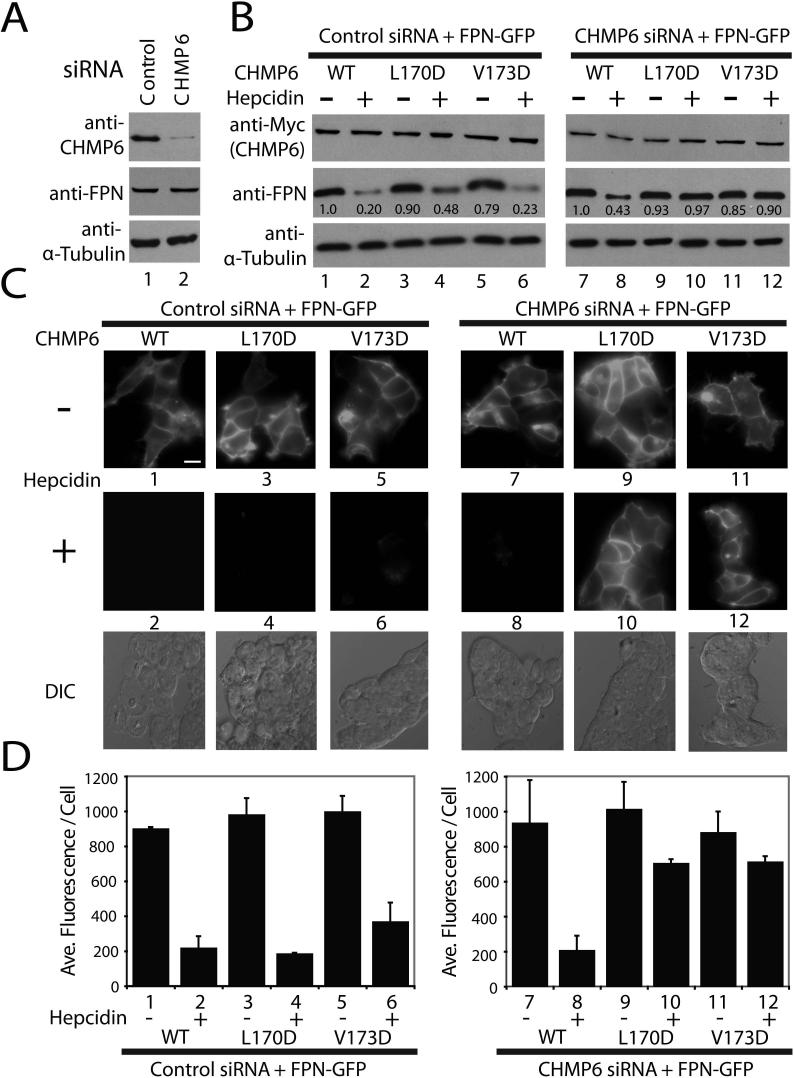
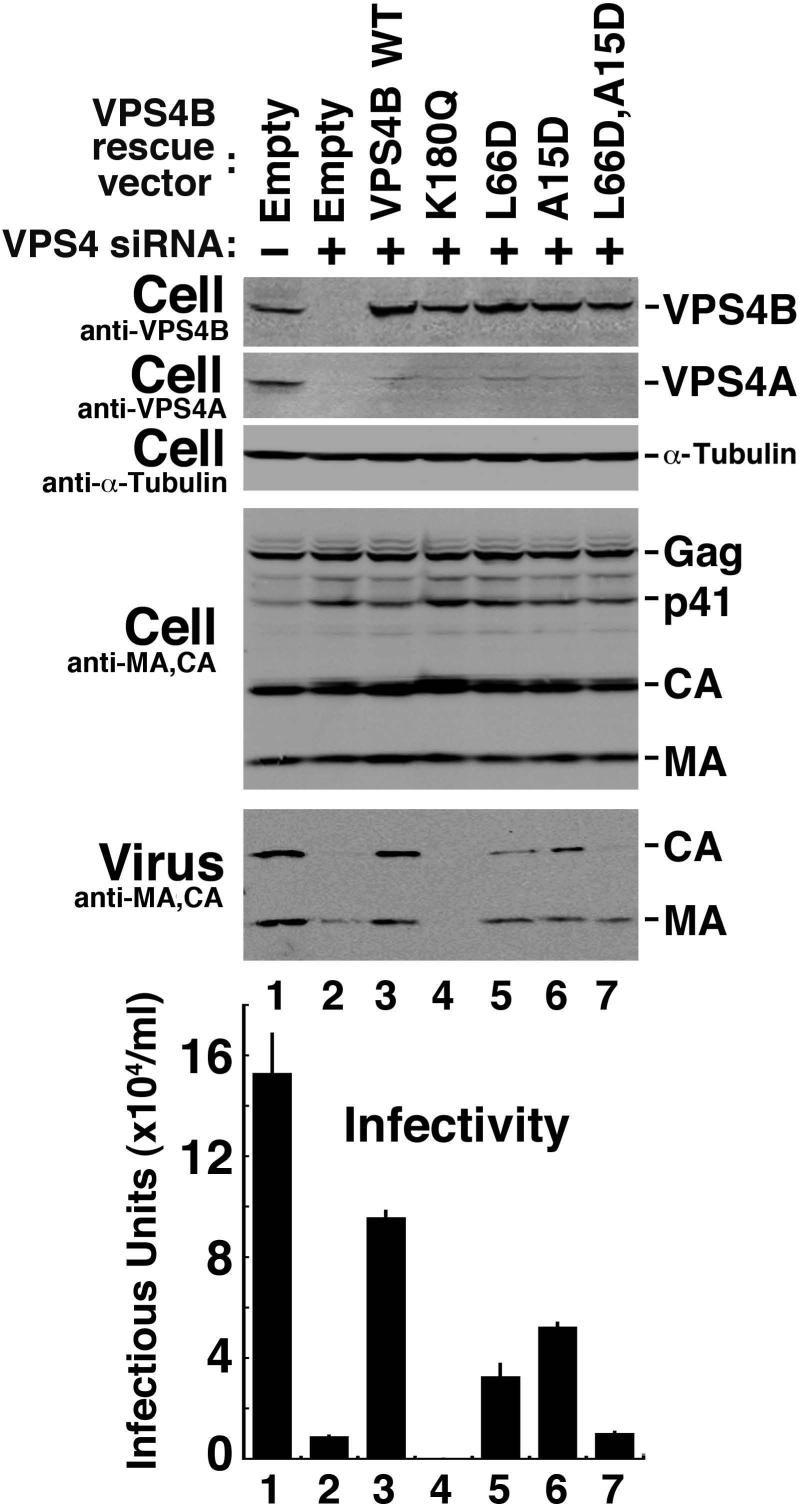
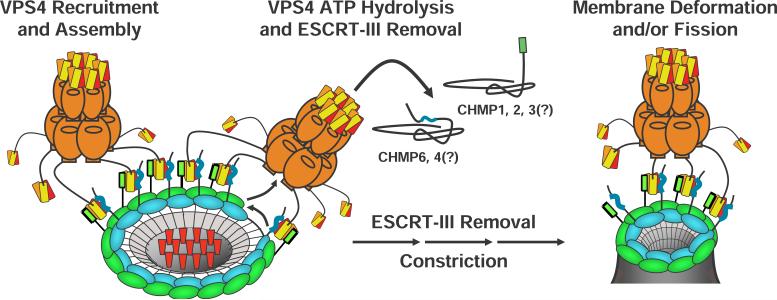
The ESCRT pathway mediates membrane remodeling during enveloped virus budding, cytokinesis, and intralumenal endosomal vesicle formation. Late in the pathway, a subset of membrane-associated ESCRT-III proteins display terminal amphipathic "MIM1" helices that bind and recruit VPS4 ATPases via their MIT domains. We now report that VPS4 MIT domains also bind a second, "MIM2" motif found in a different subset of ESCRT-III subunits. The solution structure of the VPS4 MIT-CHMP6 MIM2 complex revealed that MIM2 elements bind in extended conformations along the groove between the first and third helices of the MIT domain. Mutations that block VPS4 MIT-MIM2 interactions inhibit VPS4 recruitment, lysosomal protein targeting, and HIV-1 budding. MIT-MIM2 interactions appear to be common throughout the ESCRT pathway and possibly elsewhere, and we suggest how these interactions could contribute to a mechanism in which VPS4 and ESCRT-III proteins function together to constrict the necks of budding vesicles.
Figures
Comment in
-
Findings of Research Misconduct.Fed Regist. 2023 Sep 5;88(170):60694-60695. Fed Regist. 2023. PMID: 37736265 Free PMC article. No abstract available.
References
-
- Azmi IF, Davies BA, Xiao J, Babst M, Xu Z, Katzmann DJ. ESCRT-III Family Members Stimulate Vps4 ATPase Activity Directly or via Vta1. Dev Cell. 2008;14:50–61. - PubMed
-
- Babst M, Katzmann D, Estepa-Sabal E, Meerloo T, Emr S. Escrt-III. An endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell. 2002;3:271–282. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
