

High density lipoprotein-associated sphingosine 1-phosphate promotes endothelial barrier function
- PMID: 18606817
- PMCID: PMC2529014
- DOI: 10.1074/jbc.M801214200
High density lipoprotein-associated sphingosine 1-phosphate promotes endothelial barrier function
Abstract
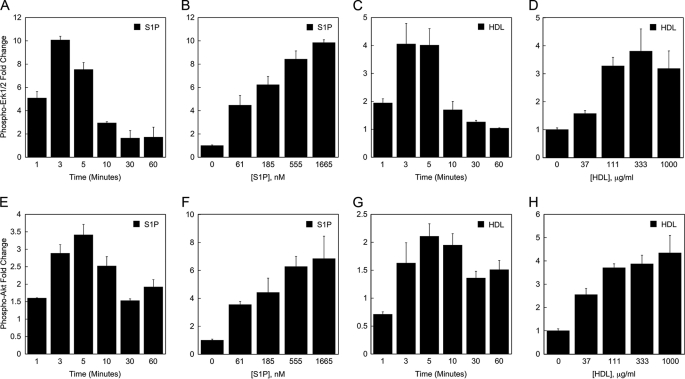
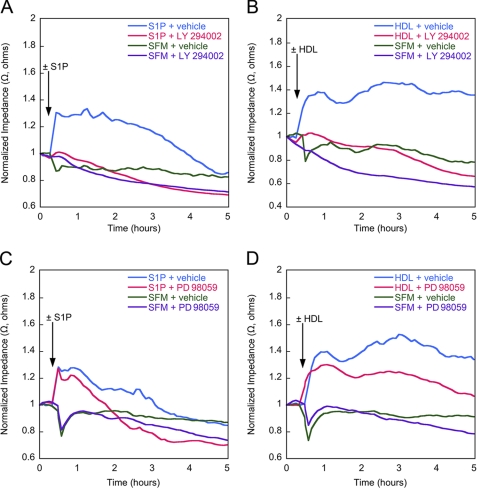
High density lipoproteins (HDL) are major plasma carriers of sphingosine 1-phosphate (S1P). Here we show that HDL increases endothelial barrier integrity as measured by electric cell substrate impedance sensing. S1P was implicated as the mediator in this process through findings showing that pertussis toxin, an inhibitor of Gi-coupled S1P receptors, as well as antagonists of the S1P receptor, S1P1, inhibited barrier enhancement by HDL. Additional findings show that HDL stimulates endothelial cell activation of Erk1/2 and Akt, signaling pathway intermediates that have been implicated in S1P-dependent endothelial barrier activity. HDL was also found to promote endothelial cell motility, a process that may also relate to endothelial barrier function in the context of a vascular injury response. The effects of HDL on endothelial cell Erk1/2 and Akt activation and motility were suppressed by pertussis toxin and S1P1 antagonists. However, both HDL-induced barrier enhancement and HDL-induced motility showed a greater dependence on Akt activation as compared with Erk1/2 activation. Together, the findings indicate that HDL has endothelial barrier promoting activities, which are attributable to its S1P component and signaling through the S1P1/Akt pathway.
Figures





References
-
- Schaphorst, K. L., Chiang, E., Jacobs, K. N., Zaiman, A., Natarajan, V., Wigley, F., and Garcia, J. G. (2003) Am. J. Physiol. 285L258 -L267 - PubMed
-
- Xu, M., Waters, C. L., Hu, C., Wysolmerski, R. B., Vincent, P. A., and Minnear, F. L. (2007) Am. J. Physiol. 4C1309 -C1318 - PubMed
-
- Peng, X., Hassoun, P. M., Sammani, S., McVerry, B. J., Burne, M. J., Rabb, H., Pearse, D., Tuder, R. M., and Garcia, J. G. (2004) Am. J. Respir. Crit. Care Med. 1691245 -1251 - PubMed
-
- Lee, J. F., Zeng, Q., Ozaki, H., Wang, L., Hand, A. R., Hla, T., Wang, E., and Lee, M. J. (2006) J. Biol. Chem. 28129190 -29200 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
