

Intrinsic disorder in scaffold proteins: getting more from less
- PMID: 18619997
- PMCID: PMC2671330
- DOI: 10.1016/j.pbiomolbio.2008.05.007
Intrinsic disorder in scaffold proteins: getting more from less
Abstract
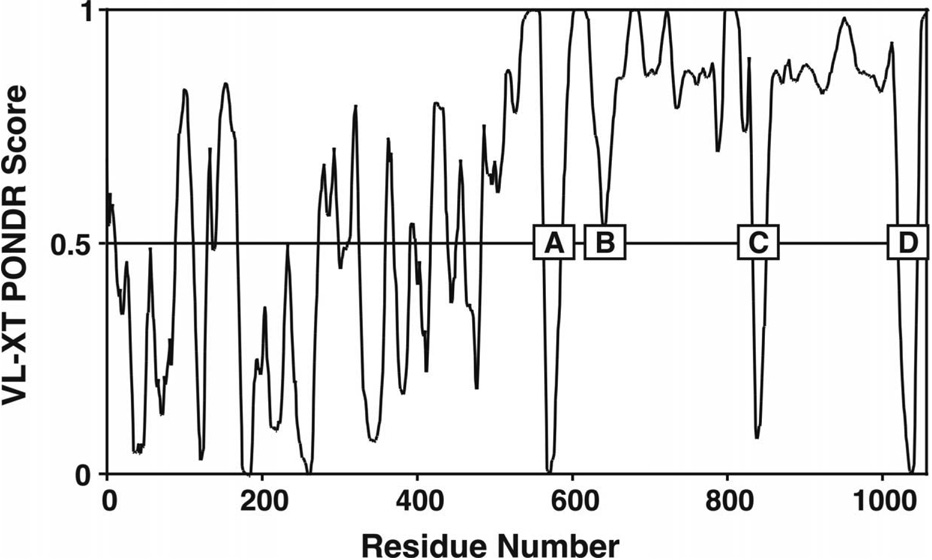
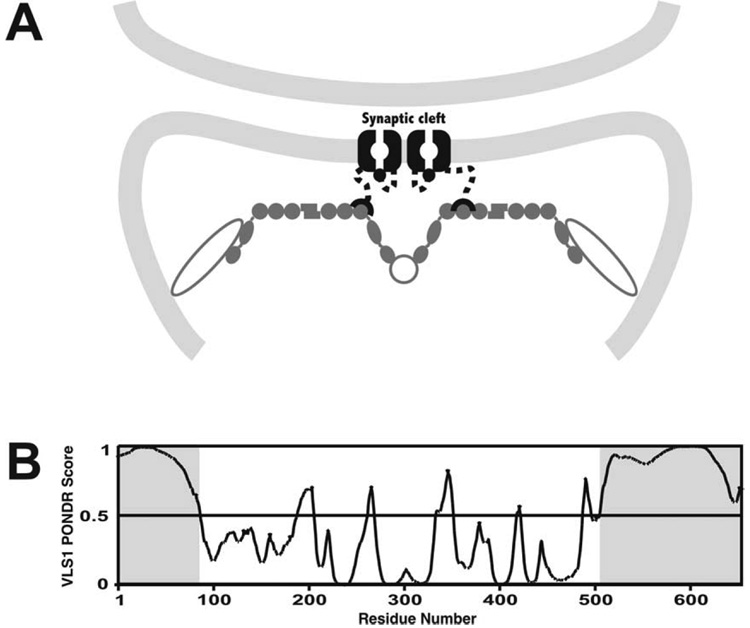
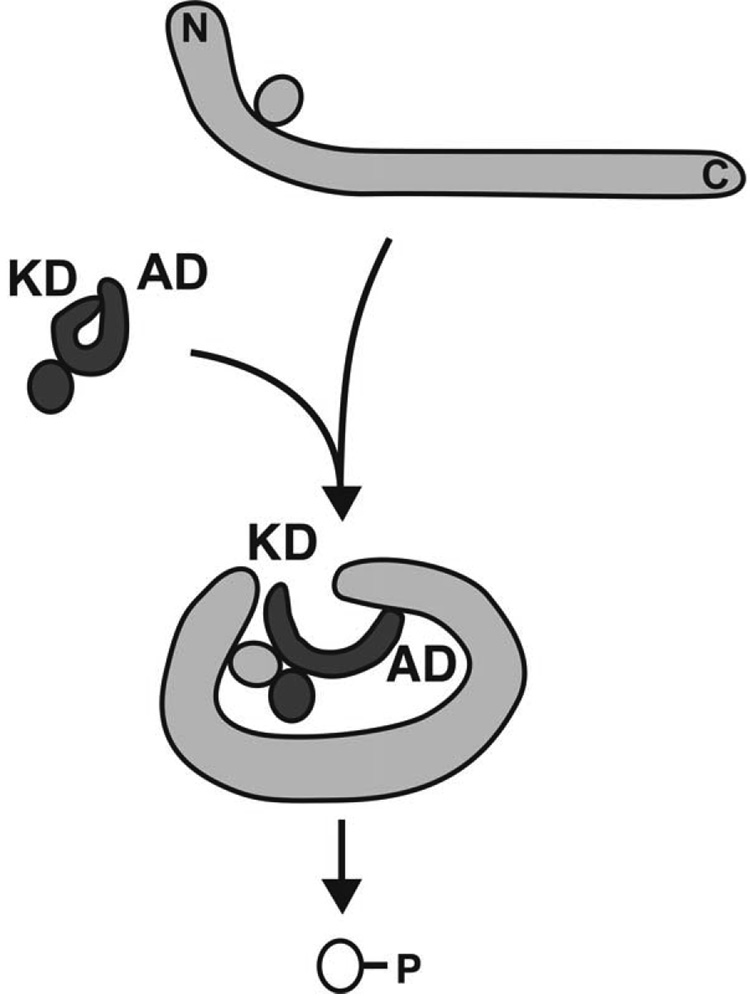
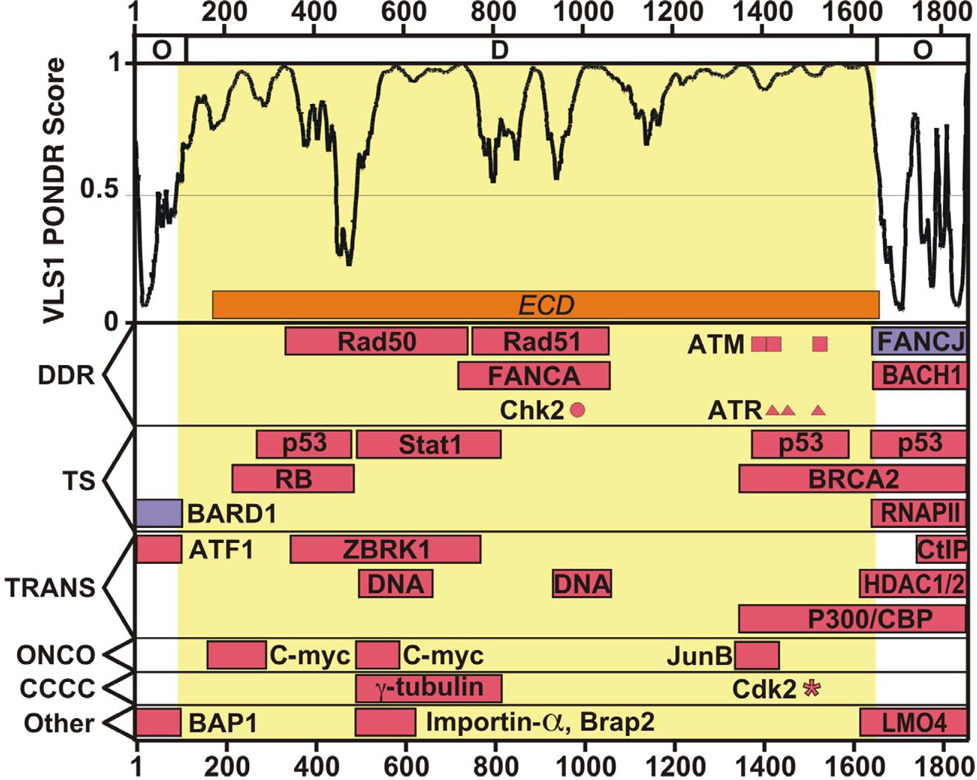
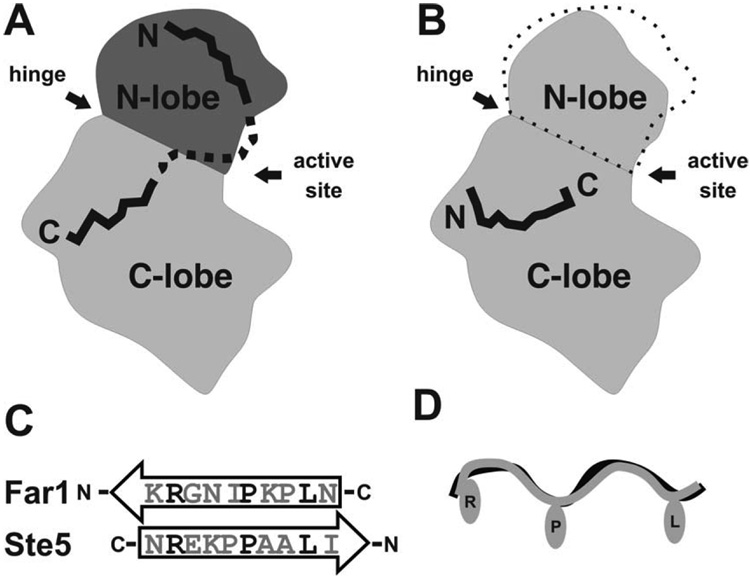
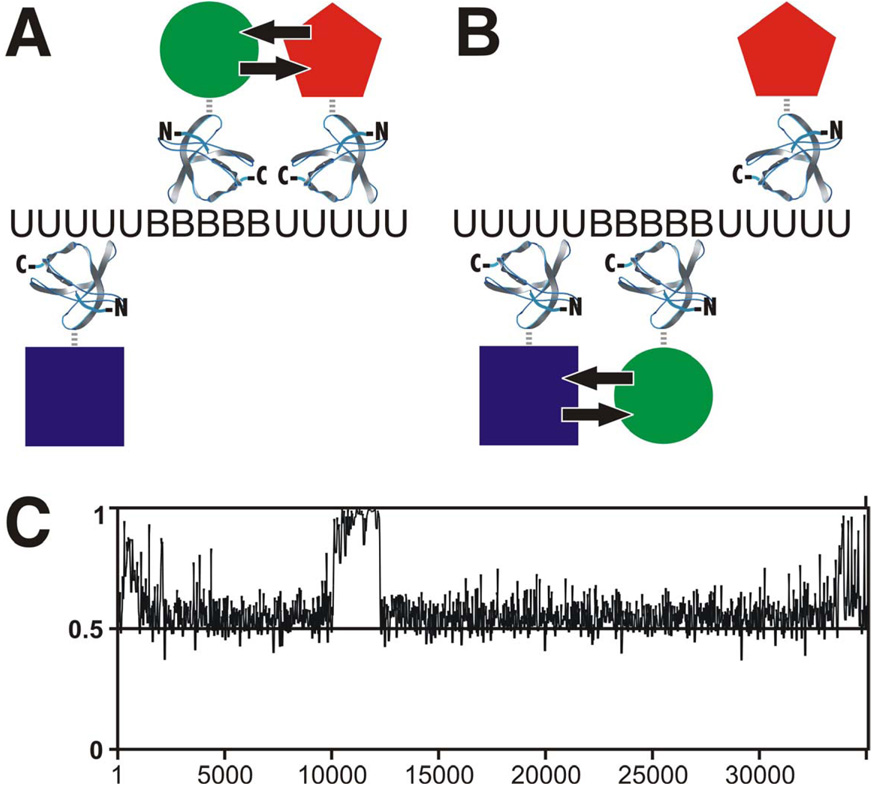
Regulation, recognition and cell signaling involve the coordinated actions of many players. Signaling scaffolds, with their ability to bring together proteins belonging to common and/or interlinked pathways, play crucial roles in orchestrating numerous events by coordinating specific interactions among signaling proteins. This review examines the roles of intrinsic disorder (ID) in signaling scaffold protein function. Several well-characterized scaffold proteins with structurally and functionally characterized ID regions are used here to illustrate the importance of ID for scaffolding function. These examples include scaffolds that are mostly disordered, only partially disordered or those in which the ID resides in a scaffold partner. Specific scaffolds discussed include RNase, voltage-activated potassium channels, axin, BRCA1, GSK-3beta, p53, Ste5, titin, Fus3, BRCA1, MAP2, D-AKAP2 and AKAP250. Among the mechanisms discussed are: molecular recognition features, fly-casting, ease of encounter complex formation, structural isolation of partners, modulation of interactions between bound partners, masking of intramolecular interaction sites, maximized interaction surface per residue, toleration of high evolutionary rates, binding site overlap, allosteric modification, palindromic binding, reduced constraints for alternative splicing, efficient regulation via posttranslational modification, efficient regulation via rapid degradation, protection of normally solvent-exposed sites, enhancing the plasticity of interaction and molecular crowding. We conclude that ID can enhance scaffold function by a diverse array of mechanisms. In other words, scaffold proteins utilize several ID-facilitated mechanisms to enhance function, and by doing so, get more functionality from less structure.
Figures


References
-
- Albert R, Jeong H, Barabasi AL. Error and attack tolerance of complex networks. Nature. 2000;406:378–382. - PubMed
-
- Andrade MA, Bork P. HEAT repeats in the Huntington's disease protein. Nat Genet. 1995;11:115–116. - PubMed
-
- Andrade MA, Perez-Iratxeta C, Ponting CP. Protein repeats: structures, functions, and evolution. J Struct Biol. 2001;134:117–131. - PubMed
-
- Aoki C, Siekevitz P. Ontogenetic changes in the cyclic denosine 3',5'-monophosphate-stimulatable phosphorylation of cat visual cortex proteins, particularly of microtubule-associated protein 2 (MAP 2): effects of normal and dark rearing and of the exposure to light. J Neurosci. 1985;5:2465–2483. - PMC - PubMed
-
- Avalos JL, Celic I, Muhammad S, Cosgrove MS, Boeke JD, Wolberger C. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol Cell. 2002;10:523–535. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
