

The immune system and cardiac repair
- PMID: 18620057
- PMCID: PMC2642482
- DOI: 10.1016/j.phrs.2008.06.007
The immune system and cardiac repair
Abstract
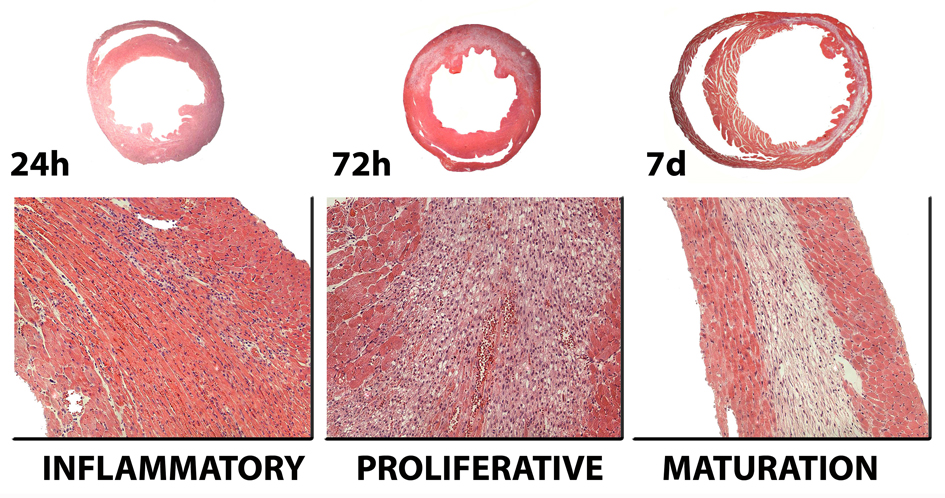
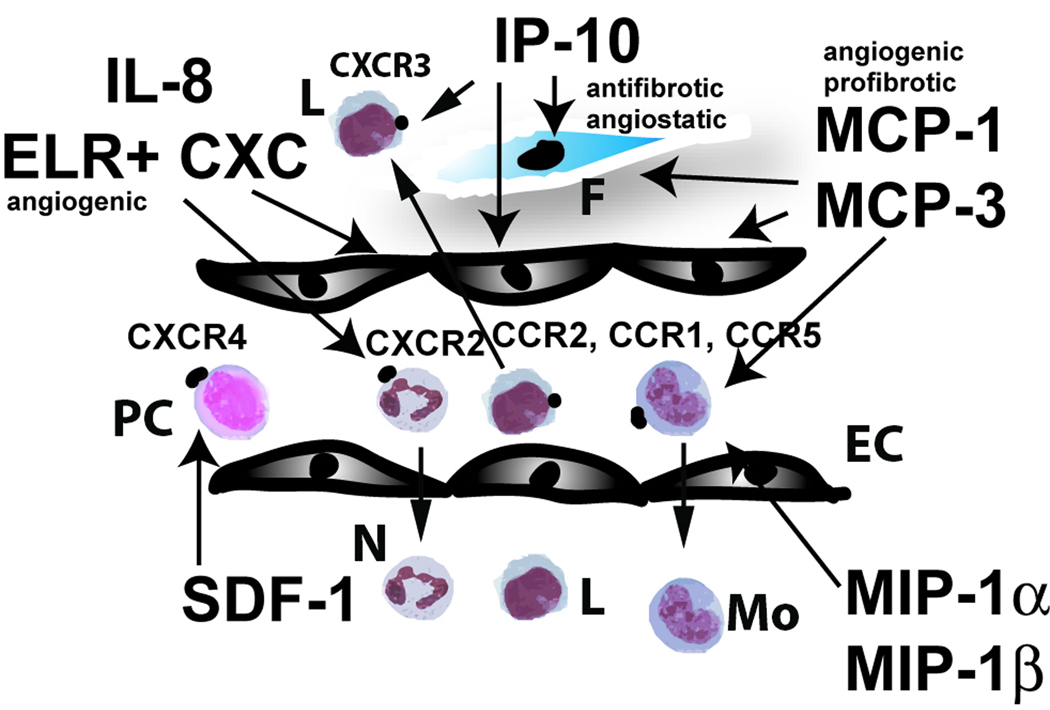
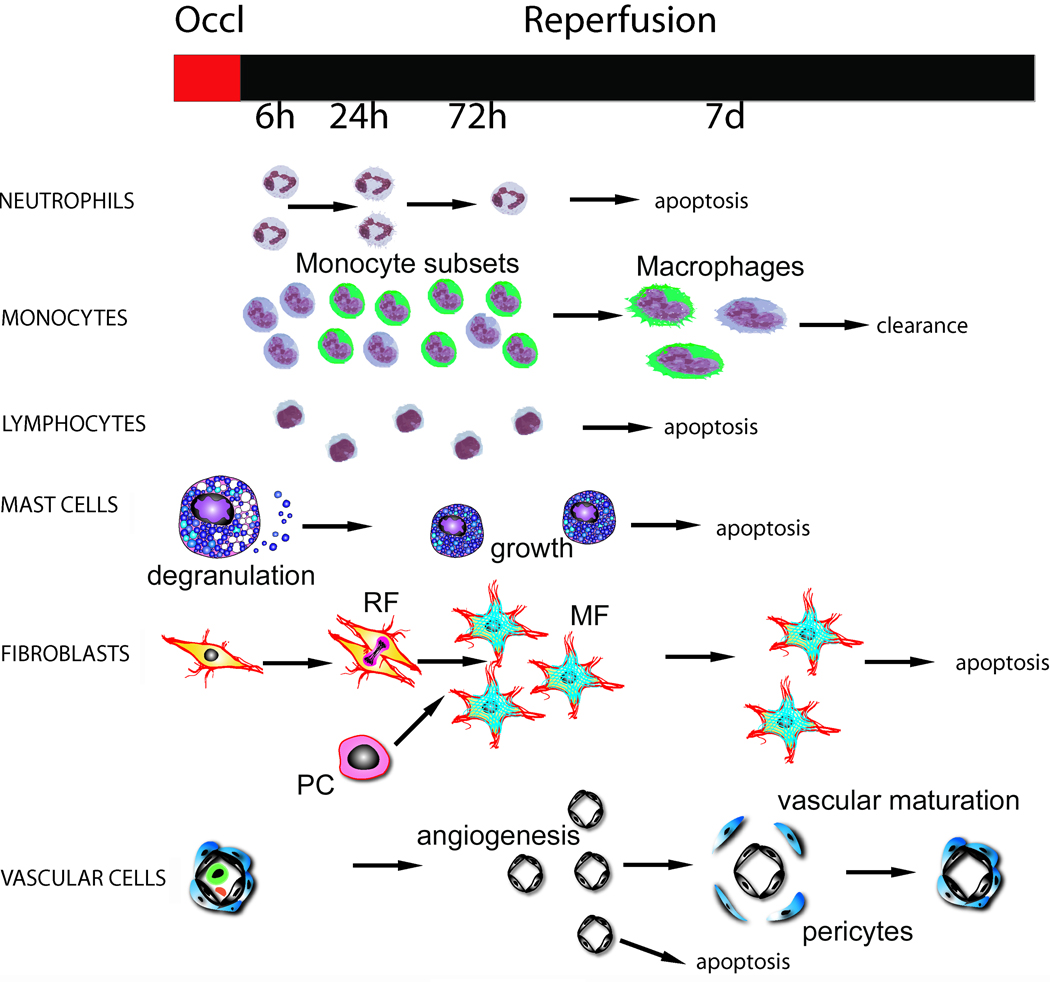
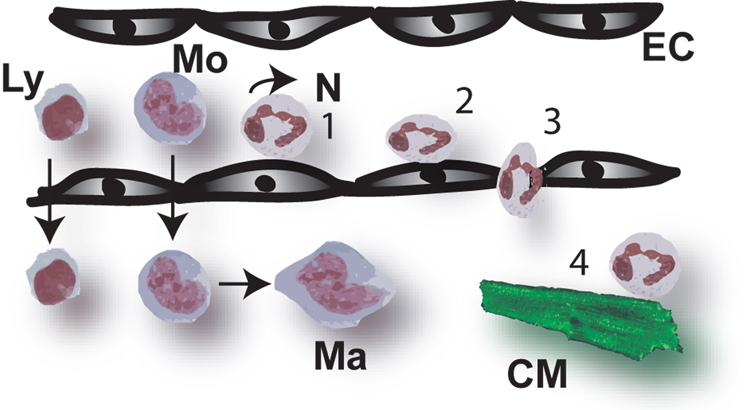
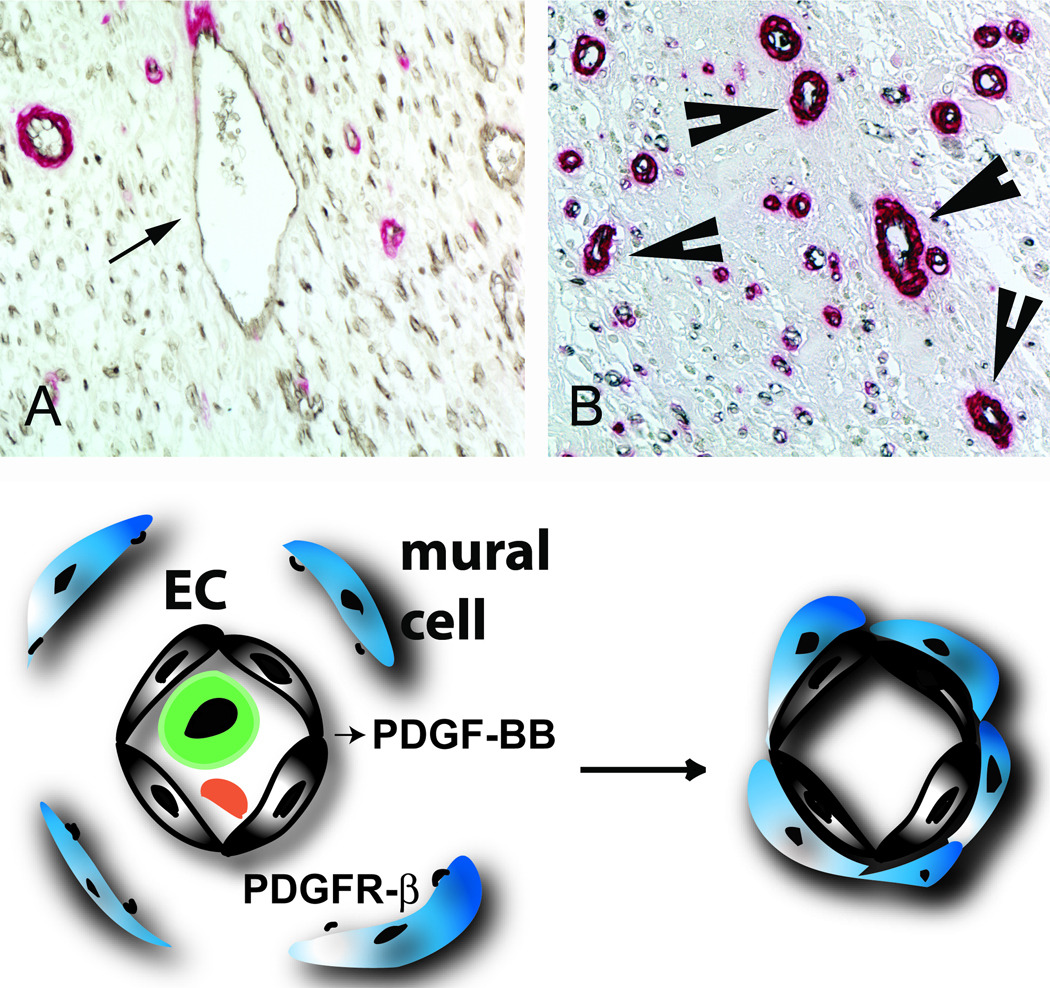
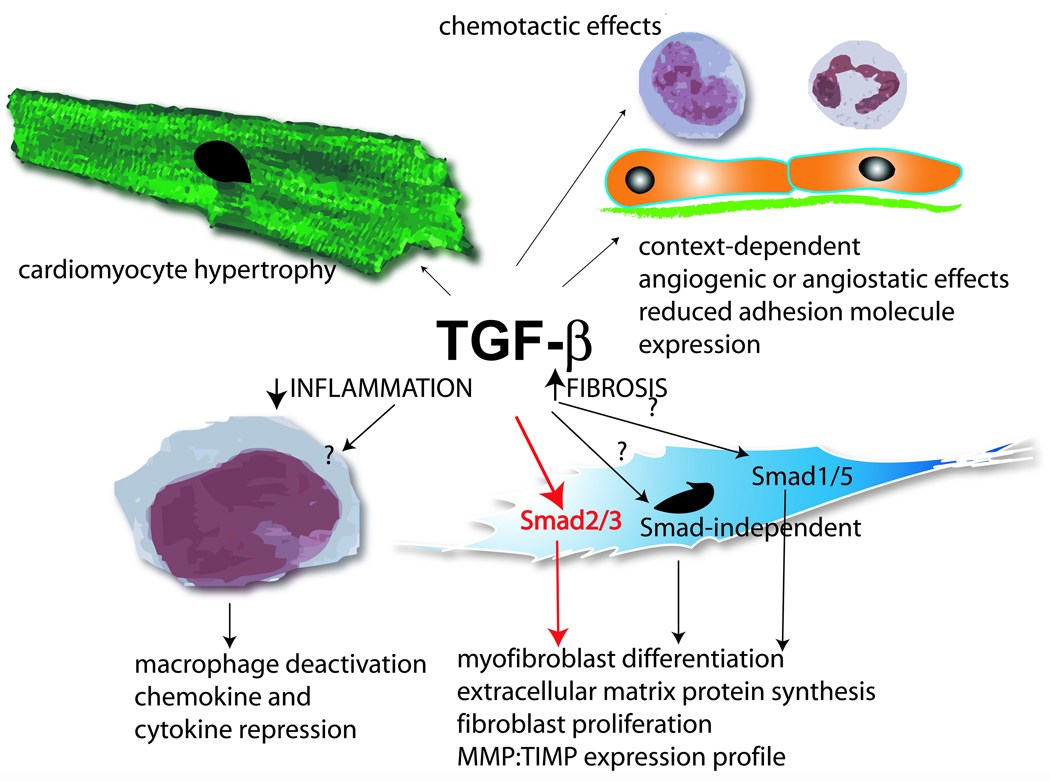
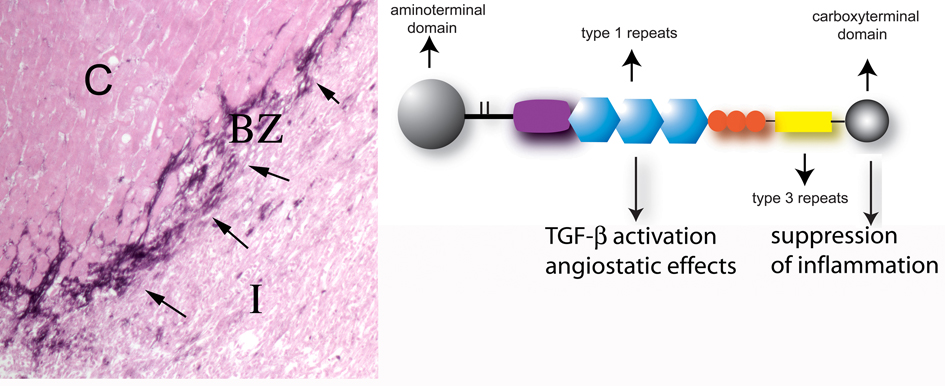
Myocardial infarction is the most common cause of cardiac injury and results in acute loss of a large number of myocardial cells. Because the heart has negligible regenerative capacity, cardiomyocyte death triggers a reparative response that ultimately results in formation of a scar and is associated with dilative remodeling of the ventricle. Cardiac injury activates innate immune mechanisms initiating an inflammatory reaction. Toll-like receptor-mediated pathways, the complement cascade and reactive oxygen generation induce nuclear factor (NF)-kappaB activation and upregulate chemokine and cytokine synthesis in the infarcted heart. Chemokines stimulate the chemotactic recruitment of inflammatory leukocytes into the infarct, while cytokines promote adhesive interactions between leukocytes and endothelial cells, resulting in transmigration of inflammatory cells into the site of injury. Monocyte subsets play distinct roles in phagocytosis of dead cardiomyocytes and in granulation tissue formation through the release of growth factors. Clearance of dead cells and matrix debris may be essential for resolution of inflammation and transition into the reparative phase. Transforming growth factor (TGF)-beta plays a crucial role in cardiac repair by suppressing inflammation while promoting myofibroblast phenotypic modulation and extracellular matrix deposition. Myofibroblast proliferation and angiogenesis result in formation of highly vascularized granulation tissue. As the healing infarct matures, fibroblasts become apoptotic and a collagen-based matrix is formed, while many infarct neovessels acquire a muscular coat and uncoated vessels regress. Timely resolution of the inflammatory infiltrate and spatial containment of the inflammatory and reparative response into the infarcted area are essential for optimal infarct healing. Targeting inflammatory pathways following infarction may reduce cardiomyocyte injury and attenuate adverse remodeling. In addition, understanding the role of the immune system in cardiac repair is necessary in order to design optimal strategies for cardiac regeneration.
Figures
References
-
- Tavazzi L. Clinical epidemiology of acute myocardial infarction. Am Heart J. 1999;138:S48–S54. - PubMed
-
- Jennings RB, Murry CE, Steenbergen C, Jr, Reimer KA. Development of cell injury in sustained acute ischemia. Circulation. 1990;82:II2–II12. - PubMed
-
- Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–1172. - PubMed
-
- Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–367. - PubMed
-
- Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–582. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
