

Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia
- PMID: 18621108
- PMCID: PMC3584171
- DOI: 10.1016/j.neuroscience.2008.06.025
Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia
Abstract
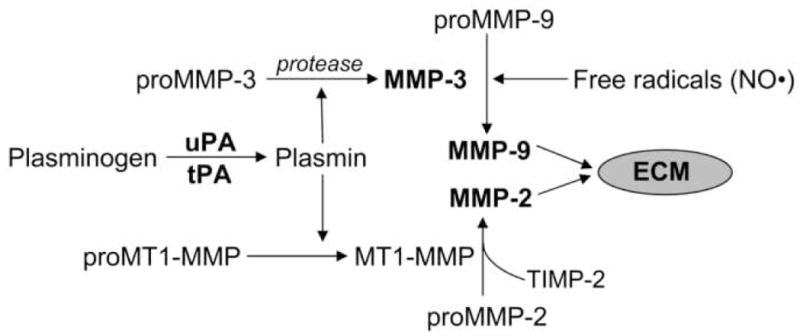
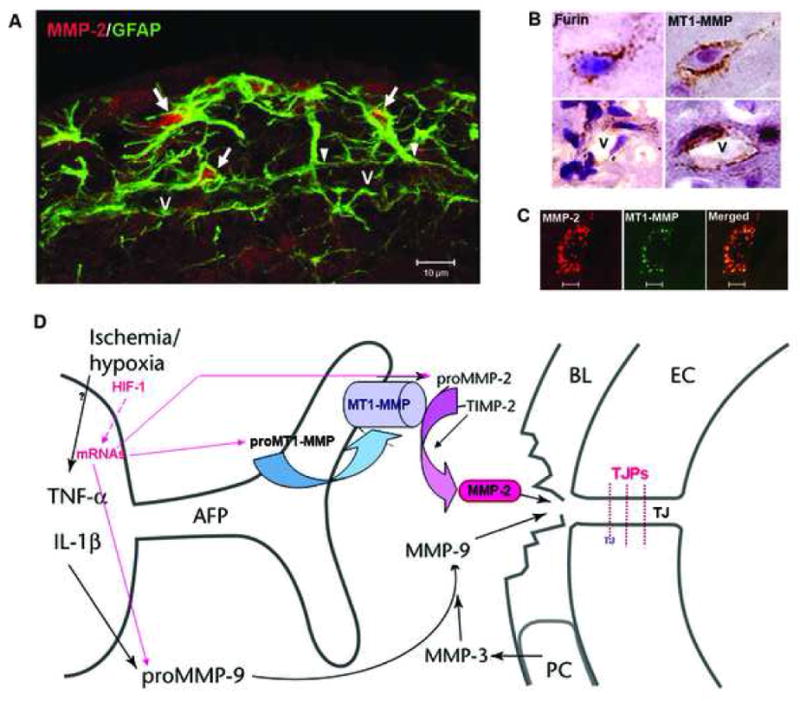
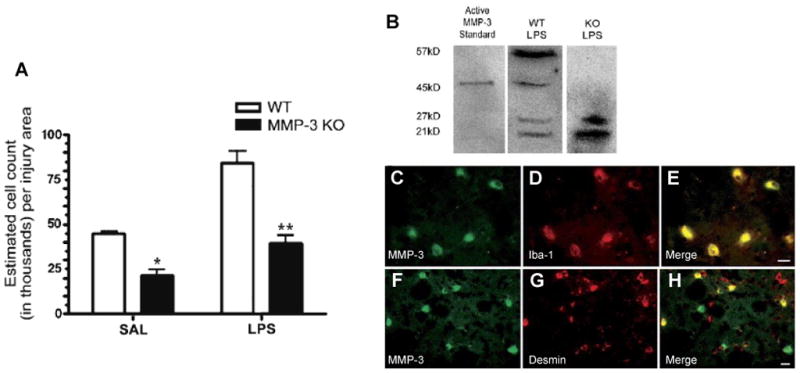
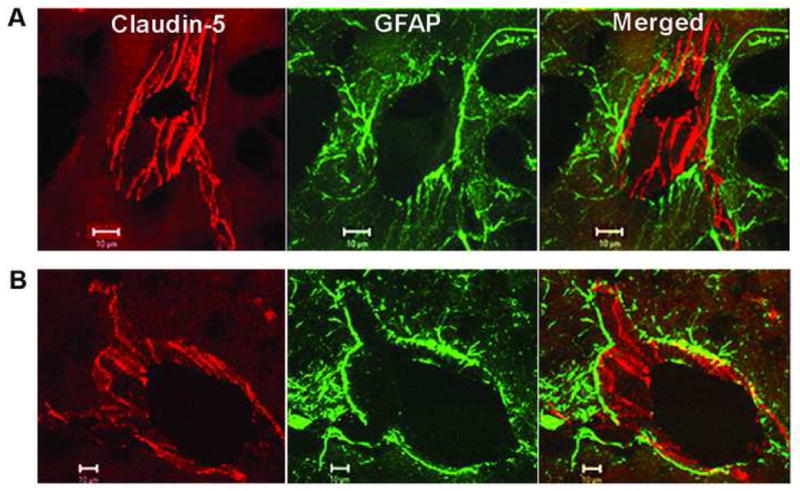
Regulation of the extracellular matrix by proteases and protease inhibitors is a fundamental biological process for normal growth, development and repair in the CNS. Matrix metalloproteinases (MMPs) and the tissue inhibitors of metalloproteinases (TIMPs) are the major extracellular-degrading enzymes. Two other enzyme families, a disintegrin and metalloproteinase (ADAM), and the serine proteases, plasminogen/plasminogen activator (P/PA) system, are also involved in extracellular matrix degradation. Normally, the highly integrated action of these enzyme families remodels all of the components of the matrix and performs essential functions at the cell surface involved in signaling, cell survival, and cell death. During the inflammatory response induced in infection, autoimmune reactions and hypoxia/ischemia, abnormal expression and activation of these proteases lead to breakdown of the extracellular matrix, resulting in the opening of the blood-brain barrier (BBB), preventing normal cell signaling, and eventually leading to cell death. There are several key MMPs and ADAMs that have been implicated in neuroinflammation: gelatinases A and B (MMP-2 and -9), stromelysin-1 (MMP-3), membrane-type MMP (MT1-MMP or MMP-14), and tumor necrosis factor-alpha converting enzyme (TACE). In addition, TIMP-3, which is bound to the cell surface, promotes cell death and impedes angiogenesis. Inhibitors of metalloproteinases are available, but balancing the beneficial and detrimental effects of these agents remains a challenge.
Figures
References
-
- Amantea D, Russo R, Gliozzi M, Fratto V, Berliocchi L, Bagetta G, Bernardi G, Corasaniti MT. Early upregulation of matrix metalloproteinases following reperfusion triggers neuroinflammatory mediators in brain ischemia in rat. Int Rev Neurobiol. 2007;82:149–169. - PubMed
-
- Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. - PubMed
-
- Asahi M, Sumii T, Fini ME, Itohara S, Lo EH. Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. Neuroreport. 2001a;12:3003–3007. - PubMed
-
- Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
