

Self-assembly of a simple membrane protein: coarse-grained molecular dynamics simulations of the influenza M2 channel
- PMID: 18621807
- PMCID: PMC2553146
- DOI: 10.1529/biophysj.108.131078
Self-assembly of a simple membrane protein: coarse-grained molecular dynamics simulations of the influenza M2 channel
Abstract
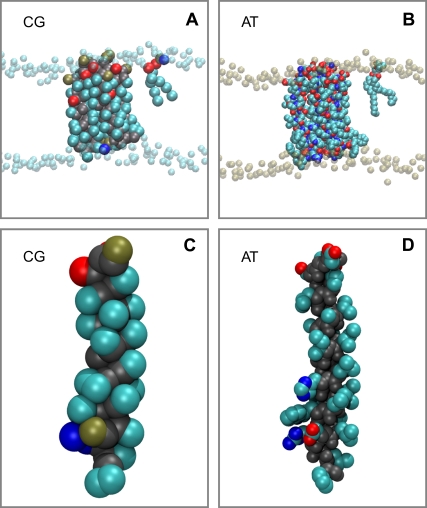
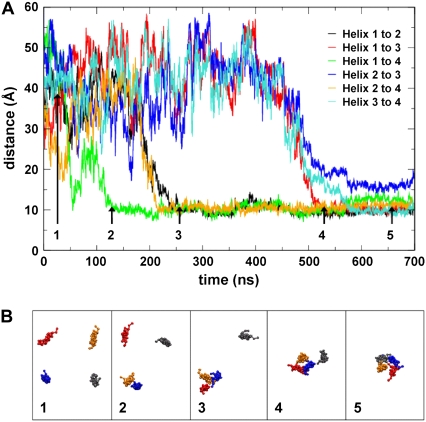
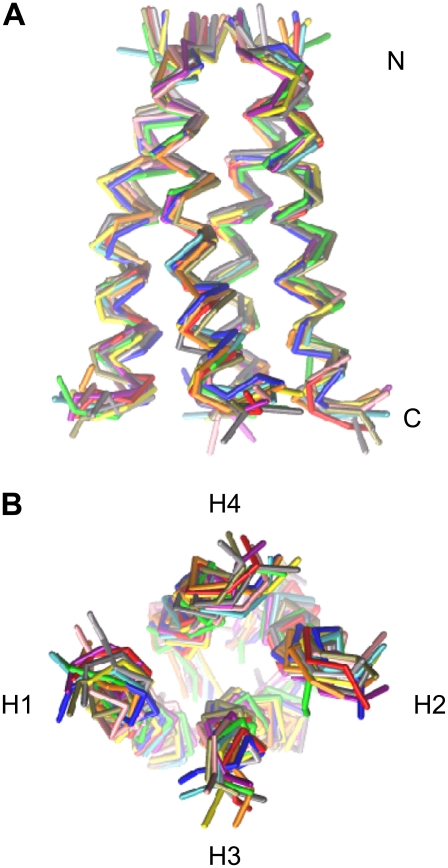
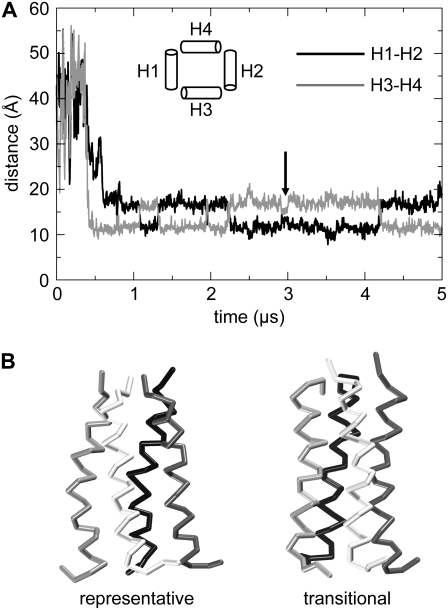
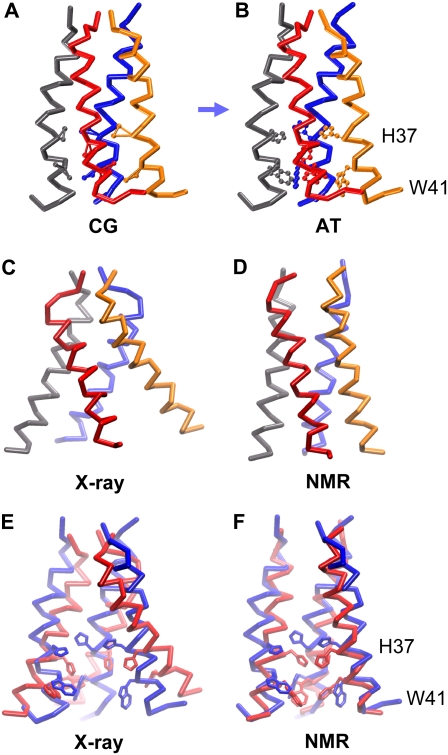
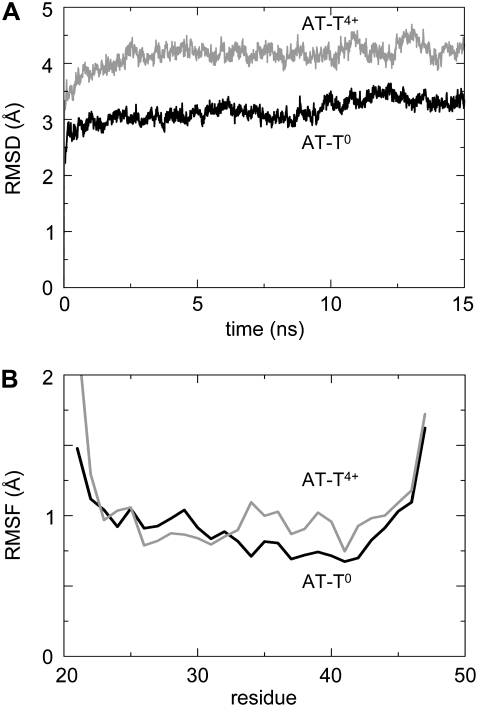
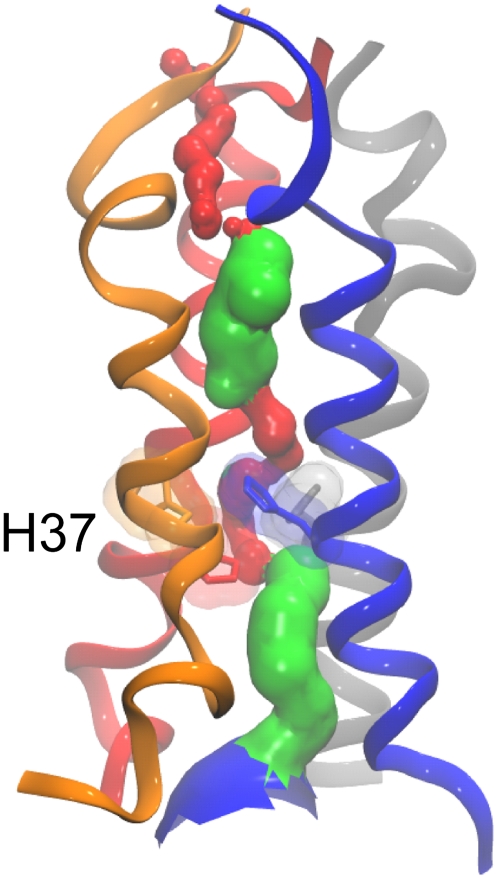
The transmembrane (TM) domain of the M2 channel protein from influenza A is a homotetrameric bundle of alpha-helices and provides a model system for computational approaches to self-assembly of membrane proteins. Coarse-grained molecular dynamics (CG-MD) simulations have been used to explore partitioning into a membrane of M2 TM helices during bilayer self-assembly from lipids. CG-MD is also used to explore tetramerization of preinserted M2 TM helices. The M2 helix monomer adopts a membrane spanning orientation in a lipid (DPPC) bilayer. Multiple extended CG-MD simulations (5 x 5 micros) were used to study the tetramerization of inserted M2 helices. The resultant tetramers were evaluated in terms of the most populated conformations and the dynamics of their interconversion. This analysis reveals that the M2 tetramer has 2x rotationally symmetrical packing of the helices. The helices form a left-handed bundle, with a helix tilt angle of approximately 16 degrees. The M2 helix bundle generated by CG-MD was converted to an atomistic model. Simulations of this model reveal that the bundle's stability depends on the assumed protonation state of the H37 side chains. These simulations alongside comparison with recent x-ray (3BKD) and NMR (2RLF) structures of the M2 bundle suggest that the model yielded by CG-MD may correspond to a closed state of the channel.
Figures

Similar articles
-
Molecular dynamics simulations of the dimerization of transmembrane alpha-helices.Acc Chem Res. 2010 Mar 16;43(3):388-96. doi: 10.1021/ar900211k. Acc Chem Res. 2010. PMID: 20017540
-
The influenza A virus M2 channel: a molecular modeling and simulation study.Virology. 1997 Jun 23;233(1):163-73. doi: 10.1006/viro.1997.8578. Virology. 1997. PMID: 9201226
-
Defining the transmembrane helix of M2 protein from influenza A by molecular dynamics simulations in a lipid bilayer.Biophys J. 1999 Apr;76(4):1886-96. doi: 10.1016/s0006-3495(99)77347-9. Biophys J. 1999. PMID: 10096886 Free PMC article.
-
Modeling the membrane environment has implications for membrane protein structure and function: influenza A M2 protein.Protein Sci. 2013 Apr;22(4):381-94. doi: 10.1002/pro.2232. Epub 2013 Mar 1. Protein Sci. 2013. PMID: 23389890 Free PMC article. Review.
-
Coarse-grained simulation: a high-throughput computational approach to membrane proteins.Biochem Soc Trans. 2008 Feb;36(Pt 1):27-32. doi: 10.1042/BST0360027. Biochem Soc Trans. 2008. PMID: 18208379 Review.
Cited by
-
Systematic multiscale simulation of membrane protein systems.Curr Opin Struct Biol. 2009 Apr;19(2):138-44. doi: 10.1016/j.sbi.2009.03.001. Epub 2009 Apr 9. Curr Opin Struct Biol. 2009. PMID: 19362465 Free PMC article. Review.
-
Mapping the druggable allosteric space of G-protein coupled receptors: a fragment-based molecular dynamics approach.Chem Biol Drug Des. 2010 Sep 1;76(3):201-17. doi: 10.1111/j.1747-0285.2010.01012.x. Epub 2010 Jul 5. Chem Biol Drug Des. 2010. PMID: 20626410 Free PMC article.
-
Modeling and simulation of ion channels.Chem Rev. 2012 Dec 12;112(12):6250-84. doi: 10.1021/cr3002609. Epub 2012 Oct 4. Chem Rev. 2012. PMID: 23035940 Free PMC article. Review. No abstract available.
-
Structure and Mechanism of the Influenza A M218-60 Dimer of Dimers.J Am Chem Soc. 2015 Dec 2;137(47):14877-86. doi: 10.1021/jacs.5b04802. Epub 2015 Aug 31. J Am Chem Soc. 2015. PMID: 26218479 Free PMC article.
-
Probing the oligomeric state and interaction surfaces of Fukutin-I in dilauroylphosphatidylcholine bilayers.Eur Biophys J. 2012 Feb;41(2):199-207. doi: 10.1007/s00249-011-0773-5. Epub 2011 Nov 11. Eur Biophys J. 2012. PMID: 22075563 Free PMC article.
References
-
- Hopkins, A. L., and C. R. Groom. 2002. The druggable genome. Nat. Rev. Drug Discov. 1:727–730. - PubMed
-
- Russ, A. P., and S. Lampel. 2005. The druggable genome: an update. Drug Discov. Today. 10:1607–1610. - PubMed
-
- Fleishman, S. J., V. M. Unger, and N. Ben-Tal. 2006. Transmembrane protein structures without X-rays. Trends Biochem. Sci. 31:106–113. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
